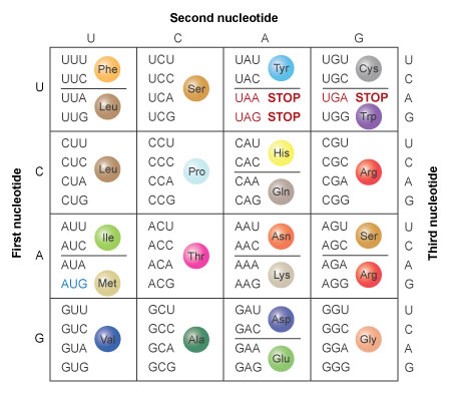
Introduction to applied bioinformatics
Osnova sekce
-
-
-
Pubmed

Search through Medline database. Full texts through FAF login.
-
Web of Science

Database Web of Science (Clavirate Analytics) includes bibliographic materials from leading scientific journals from all fields. Enables managing of references through "WebEndNote".
-
-
-
Human protein atlas
Protein databases / sequences retrieval:
Expasy / UniProt

Expasy is Swiss bioinformatics resource portal providing access to databases and software tools from a range of life science including genomics, proteomics, system biology etc.
Uniprot
 is high-quality and freely accessible resource of protein sequence and functional information.
is high-quality and freely accessible resource of protein sequence and functional information.
detail tutorial:NCBI protein
 "National Center for Biotechnology Information Protein" The Protein database is a collection of sequences from several sources, including translations from annotated coding regions.
"National Center for Biotechnology Information Protein" The Protein database is a collection of sequences from several sources, including translations from annotated coding regions.
-
-
-
This (.ab1) is "unsupported" format, needs to be saved and then opened in chromas.
-
-