Patogeneze ATS
Patogeneze ATS není zcela objasněna, ale prvotním krokem v jejím vzniku by pravděpodobně mohlo být poškození endotelu (chronické mechanické nebo chemické poškození) a odpověď na ně by mohla posléze vést ke vzniku plaků.
Fyziologická úloha endotelu a endoteliální dysfunkce
Endotel (vnitřní výstelka cévní stěny) je tvořen jednou vrstvou buněk a zastává celou řadu fyziologických funkcí, jako jsou regulace krevního tlaku, permeability a hemostázy či interakce s buňkami v krevním oběhu. Strukturní a funkční integrita endotelu je nezbytná pro udržení homeostázy cévní stěny a krevní cirkulace. Endotelové buňky tvoří semipermeabilní membránu, která kontroluje permeabilitu, tedy přenos buněčných i nebuněčných komponent krve do cévní stěny a okolních tkání. Nebuněčné složky krve jsou přenášeny pomocí transcelulárních i paracelulárních transportních mechanismů, zatímco přenos buněčných komponent je realizován pomocí různých adhezivních molekul, které jsou exprimovány na povrchu endotelu i na povrchu buněk (viz. kapitola Zánět). Endotel kontroluje optimální průtok krve cévou díky syntéze vasoaktivních látek. Mezi nejvýznamnější vasoaktivní látky patří oxid dusnatý a prostacyklin (PGI2), jejichž účinky se vzájemně potencují. Mají mohutný vasodilatační účinek, inhibují expresi adhezních molekul, adhezi a agregaci trombocytů a apoptosu endoteliálních buněk. Brzdí též migraci hladkosvalových buněk cév (VSMCs) z médie, jejich proliferaci a transformaci v sekreční buňky, čímž stabilizují strukturu cévní stěny. K zachování rovnováhy produkuje endotel též vasokonstrikčně působící faktory endotelin-1 a tromboxan A2 (TxA2), jejichž plný vasokonstrikční účinek se však projeví až při dysfunkci endotelu. Zdravý endotel je dokonale nesmáčivým povrchem, který reguluje integritu cévního řečiště. Díky expresi trombomodulinu a vazbě antitrombinu III na svém povrchu vykazuje endotel antiagregační vlastnosti. Endotel rovněž ovlivňuje fibrinolýzu, protože syntetizuje jak tkáňový aktivátor plasminogenu (tPA), který aktivuje plasminogen na plasmin štěpící fibrinová koagula, tak inhibitor aktivátoru plasminogenu-1 (PAI-1), který reguluje aktivitu tPA. Obě tyto sloučeniny jsou v endotelu syntetizovány a v různém poměru uvolňovány.
Soubor změn endoteliálních funkcí, které vznikají před začátkem strukturálních atherosklerotických změn, se označuje jako dysfunkce endotelu. Jedná se o funkční poškození endotelu, které je charakterizované zvýšenou permeabilitou cévní stěny a nerovnováhou mezi vasoaktivními, hemokoagulačními a mitogenními faktory, což vede k poruše relaxace cév, podpoře agregace trombocytů, zvýšení proliferace hladké svaloviny cév a adhezi leukocytů k povrchu endotelových buněk. Dysfunkce endotelu je vlastně kompenzační reakcí na vzniklé poškození. Dysfunkce endotelu je vratným presymptomatickým stádiem ATS a může být způsobena buď funkčním poškozením endotelu při zachování jeho morfologické integrity (např. působení virů, bakterií, toxinů, homocysteinu či LDL), nebo mechanickým poškozením, které způsobuje jeho morfologické změny (např. arteriální hypertenze). Pokud není příčina dysfunkce odstraněna, dochází ke zvýšenému pronikání atherogenních lipoproteinů do subendotelového prostoru a ke snížení tvorby PGI2 a NO, které potlačuje proliferaci buněk. Z membrán poškozeného endotelu se uvolňuje faktor aktivující destičky (PAF), který má prozánětlivé a proagregační účinky.
Dysfunkční endotel umožňuje zvýšený průnik peptidů a atherogenních lipoproteinů (obsahují apoB100) krevní plasmy do subendotelového prostoru (Obr. 2). Aktivace endotelu je provázena produkcí adhezních molekul, cytokinů a růstových faktorů, které přitahují do místa poškození monocyty, které se mění na makrofágy a pohlcují oxLDL a oxidovaný cholesterol za vzniku pěnových buněk. Ty nejsou schopny migrovat zpátky do krevního oběhu a v subendotelovém prostoru se rozpadají. Uvolněné lipidy se stávají základem lipidového jádra atheromového plátu. Makrofágy a pěnové buňky přitahují do subendotelu neutrofily a T‑lymfocyty, což podporuje rozvoj chronického zánětu. Následně dojde ke zvýšení migrace VSMCs z medie do intimy a jejich proliferace, k tvorbě mezibuněčné hmoty (ECM) a ztluštění intimy, které je kompenzováno vasodilatací kvůli zachování průsvitu cévy. V pozdějších fázích procesu dochází k vaskularizaci plátu. Tato zvýšená angiogenní aktivita je známkou destabilizace plátu, ke které dochází díky aktivaci matrix-metaloproteinas (MMP, produkované pěnovými buňkami), které rozkládají fibrózní čepičku nad lipidovým jádrem. Do plátu se mohou ukládat vápenaté soli, dochází tak k jeho kalcifikaci. Povrch plátu se stává díky působení chemokinů trombogenním a je náchylný k ruptuře.
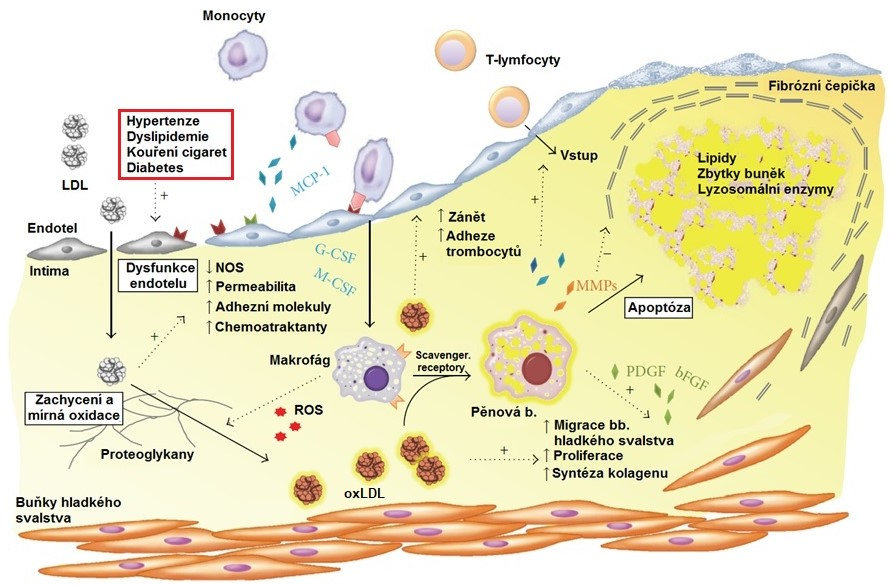
Obr. 2. Úloha oxidovaných LDL v patogenezi atherosklerosy (upraveno z Maiolino et al. 2013)
Infiltrace, retence a modifikace LDL
Nejvýznamnějším rizikovým faktorem ATS jsou proatherogenní lipoproteiny, zejména LDL, které obsahují apoB100. LDL částice jsou v plasmě zastoupeny čtyřmi frakcemi s různou velikostí a hustotou. Frakce III a IV, které jsou označovány jako malé husté LDL (sdLDL, „small dense LDL“), obsahují méně cholesterolu a mají výraznější proatherogenní vlastnosti. Díky své velikosti snadno pronikají do subendotelového prostoru, jsou snáze oxidované a vlivem odlišné konformace apoB100 mají nižší afinitu k LDL receptoru (díky tomu jsou pomaleji odstraňovány z plasmy) a jsou odstraňovány pomocí scavengerových receptorů makrofágů. Zvýšená koncentrace sdLDL v plasmě bývá provázena hypertriacylglycerolemií a sníženou hladinou HDL. Tato metabolická triáda bývá označována jako fenotyp B velikosti LDL a je nejčastějším metabolickým rizikovým faktorem zjištěným u osob s předčasnou ischemickou chorobou srdeční, kteří mají normální nebo mírně zvýšené hladiny celkového cholesterolu a LDL cholesterolu. V populaci je však častější fenotyp A, u kterého převažují frakce LDL I a II (viz kapitola 8 – Metabolismus lipoproteinů).
Určitá část LDL částic, které proniknou do subendotelového prostoru, zde zůstávají zachyceny na řetězcích proteoglykanů ECM a jsou působením RONS oxidovány. K retenci LDL dochází díky elektrostatické interakci mezi kladně nabitými aminokyselinami několika domén apoB100 a záporně nabitými řetězci glykosaminoglykanů v molekulách proteoglykanů ECM (např. biglykan, versikan). Zachycené LDL částice jsou náchylné k modifikaci různými biologicky aktivními látkami, po níž se stávají proatherogenními. Mezi popsané modifikace patří oxidace, glykace, agregace/fúze, tvorba komplexů s proteoglykany, vznik imunokomplexů s protilátkami a reakce s homocysteinem. K oxidaci LDL tedy s velkou pravděpodobností nedochází v krevním oběhu díky antioxidačním vlastnostem krve. Místem vzniku oxLDL v podmínkách in vivo je subendotelový prostor. V plasmě jsou však oxLDL detekovatelné, část oxLDL může být totiž díky obousměrnému transportu přenesena zpět do krevní cirkulace.
Oxidace LDL (Obr. 3) probíhá v subendotelovém prostoru a je vyvolána enzymy, které pocházejí z makrofágů, endotelových buněk a VSMCs. Jedná se např. o myeloperoxidasu, lipoxygenasy či NADPH-oxidasu. K oxidaci LDL může docházet rovněž vlivem ROS a iontů přechodných kovů. Působením ROS, iontů přechodných kovů a lipoxygenas dochází především k oxidaci lipidových složek LDL, kdy vznikají oxysteroly z cholesterolu, lipoperoxidy, isoprostany a ketoaldehydy z mastných kyselin a oxidované fosfolipidy. Vznikají tzv. minimálně modifikované LDL (mmLDL), které mají modifikovanou lipidovou část molekuly a změny ve struktuře apoB100 jsou malé. Díky tomu jsou mmLDL odstraňovány LDL receptorem a mají jen slabé atherogenní vlastnosti. Přesto jsou schopné indukovat v endotelových buňkách syntézu chemoatraktantů pro monocyty (MCP-1), faktoru stimulujícího kolonie monocytů (M-CSF) a adhezních molekul pro monocyty (VCAM-1, ICAM-1). Myeloperoxidasa modifikuje hlavně proteinovou část molekuly LDL za vzniku silně oxidovaných LDL (oxLDL). Oxidace apoB100 je provázena oxidací aminokyselinových zbytků (chloro- a nitrotyrosiny), fragmentací řetězce proteinu a tvorbou zkřížených vazeb („cross-linking“). Následkem oxidace apoB100 nejsou oxLDL rozpoznány LDL receptorem a jsou odstraňovány scavengerovými receptory makrofágů, ze kterých se následně stávají pěnové buňky. oxLDL mají silné atherogenní vlastnosti, indukují v makrofázích syntézu cytokinů a růstových faktorů, působí cytotoxicky na endotelové buňky a VSMCs, narušují vasorelaxaci inhibicí NO-synthasy, vyvolávají dělení VSMCs a makrofágů, podporují agregaci trombocytů, snižují fibrinolytickou aktivitu a působí imunogenně (navozují tvorbu protilátek a aktivaci T-lymfocytů).
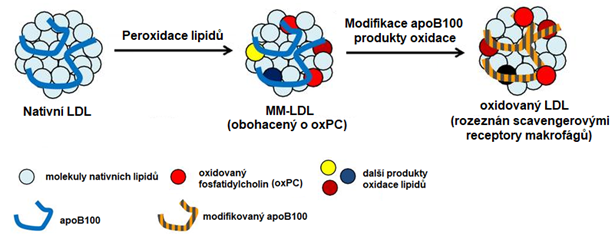
Obr. 3. Vznik minimálně modifikovaných a oxidovaných LDL částic (upraveno z Itabe et al. 2011)
Dalším typem modifikace popsané u LDL je jejich glykace (AGE-LDL), tj. vazba glukosy na apoB100 a následná tvorba koncových produktů pokročilé glykace (AGEs; viz. kapitola Poruchy regulace glykemie, diabetes mellitus). U diabetiků byly popsány zvýšené hladiny AGE-LDL než u zdravých osob. K tvorbě AGE produktů dochází zejména na kladně nabitých lysinových zbytcích ve vazebné doméně pro LDL receptor, což vyvolá snížení afinity LDL receptoru k AGE-LDL. AGE-LDL jsou náchylnější k oxidační modifikaci (Obr. 4).
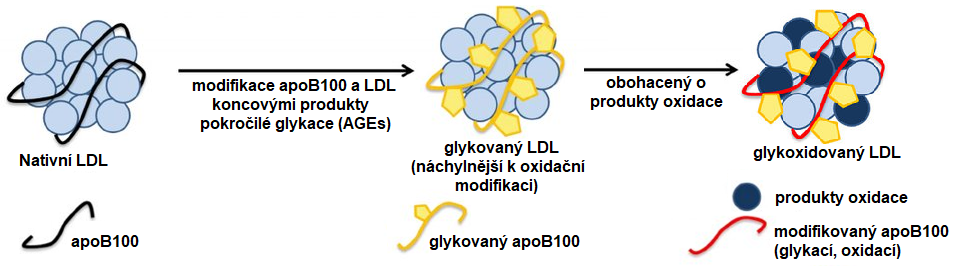
Obr. 4. Vznik glykovaného a glykoxidovaného LDL (upraveno z Alique et al. 2015)
Během posledních let byla v plasmě pacientů s ATS popsána rovněž desialylace LDL částic (katalyzovaná trans-sialidasou) a vznik elektronegativních LDL částic (LDL(-)), které jsou pravděpodobně příčinou vzniku velkých atherogenních imunokomplexů. LDL(-) mají vyšší tendenci k agregaci a jejich přítomnost v plasmě je spojena s vyšším rizikem vzniku kardiovaskulárních onemocnění. Vznik LDL(-) je pravděpodobně výsledkem série změn LDL částic, které zahrnují desialylaci, ztrátu lipidů, zmenšení velikosti a peroxidaci, probíhajících v plasmě (Obr. 5). LDL(-) mají vyšší atherogenní potenciál než nativní LDL. Tyto částice mohou aktivovat imunitní odpověď organismu (včetně tvorby autoprotilátek) a tak přispívat k progresi ATS. Jsou náchylnější ke shlukování a tvorbě komplexů, mají delší biologický poločas, díky menší velikosti snáze pronikají do subendotelového prostoru, ve zvýšené míře se vážou na proteoglykany ECM. U těchto částic je pozměněné prostorové uspořádání apoB-100 a díky tomu nejsou tyto částice schopné interakce s LDL-receptorem a jsou odstraňovány scavengerovými receptory makrofágů.
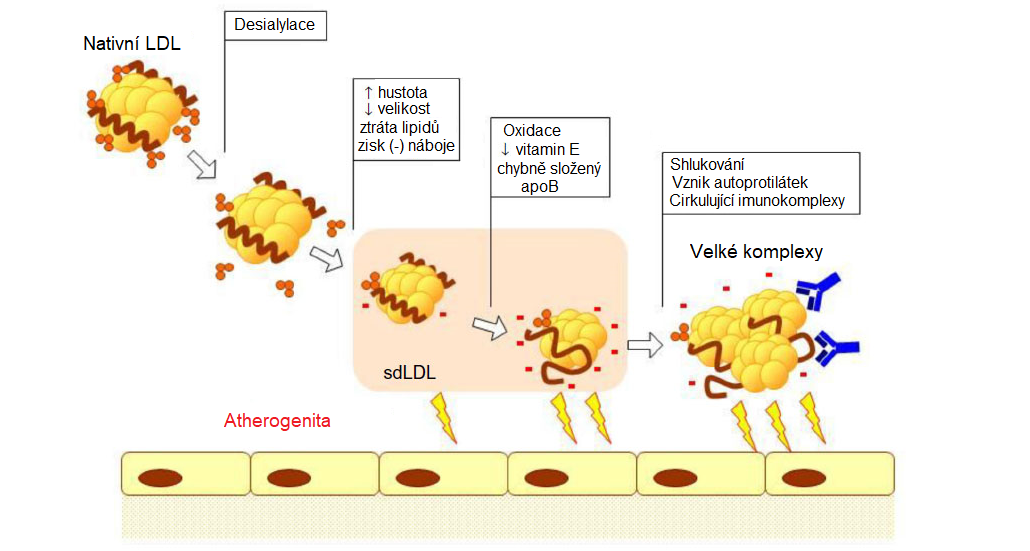
Obr. 5 Atherogenní modifikace LDL v plasmě (upraveno z Ivanova et al. 2015)
Nábor, transmigrace a diferenciace leukocytů vyvolaná částicemi LDL
Modifikované LDL částice indukují v endotelových buňkách sekreci chemotaktických látek pro monocyty a T-lymfocyty (např. MCP-1), které tyto buňky přitahují do místa poškození, a expresi adhezních molekul (zejm. VCAM-1), které jim umožňují adhezi k endotelu a průnik arteriální stěnou. V intimě cév se monocyty vlivem růstových faktorů (M-CSF) transformují do makrofágů a exprimují na svém povrchu scavengerové receptory, zajišťující příjem modifikovaných LDL částic. Makrofágy se mění na pěnové buňky, které jsou charakteristickou buněčnou složkou atherosklerotických lézí. Pěnové buňky produkují cytokiny, růstové faktory, matrix-metaloproteinasy, ROS a tkáňový faktor, které podporují zánětlivou odpověď včetně přestavby cévní stěny, zvýšené náchylnosti plaků k ruptuře a následnému vzniku trombů. Pěnové buňky (ale i endotelie) produkují např. destičkový růstový faktor (PDGF) a bazický fibroblastový růstový faktor (bFGF), které indukují růst a dělení pojivových buněk (VSMCs, fibroblasty) a syntézu ECM.
Zatím je známo 11 tříd scavengerových („zametacích“) receptorů makrofágů. Tyto povrchové receptory rozeznávají celou řadu ligandů (nízká specifičnost) a přispívají k odstraňování cizorodých nebo pozměněných vlastních struktur. Odstraňování ligandů probíhá endocytózou. Podobnost primární struktury těchto proteinů je malá nebo žádná, jejich společnou vlastností je, že jejich exprese není řízena intracelulární koncentrací cholesterolu (na rozdíl od LDL-receptoru) a může tedy docházet k jeho hromadění uvnitř makrofágů. Na internalizaci modifikovaných LDL částic se podílejí zejména SR-AI/II, CD-36 (též SR-BI) a LOX-1 (též SR-E1, „lectin-type oxidized LDL receptor-1“). SR-AI rozeznává řadu ligandů, z lipidových a lipoproteinových ligandů jsou to např. lysofosfatidylcholin, fosfatidová kyselina, cholesterol, ox-LDL, acetylované LDL a MDA-LDL (MDA – malondialdehyd, produkt peroxidace lipidů). Spolu s CD36 jsou hlavními receptory, které přijímají a odstraňují 75-90 % modifikovaných LDL částic. Vazba ox-LDL na CD36 spouští v makrofázích prozánětlivé signální dráhy, jejichž výsledkem je aktivace transkripčního faktoru NF-κB a následná syntéza prozánětlivých cytokinů IL-1 a TNF-α. Interakce oxLDL-CD36 navíc vyvolává inhibici migrace makrofágů, které se tak hromadí v atherosklerotických lézích. LOX-1 je schopný vázat různé modifikované LDL (např. ox-LDL, AGE-LDL), ale i apoptotické/senescentní buňky, kardiolipin, AGEs, aktivované destičky a další ligandy.
T-lymfocyty, které se dostaly do ATS plaku, produkují po aktivaci antigenem (např. ox-LDL, proteiny teplotního šoku) cytokiny, které ovlivňují chování dalších buněk. Rovněž dochází k interakci povrchových molekul T-lymfocytů a makrofágů (interakce mezi CD154 a CD40), jejímž výsledkem je zvýšení produkce cytokinů, MMP a tkáňových faktorů makrofágy.
Přestavba cévní stěny
Za normálního stavu produkují VSMCs v medii cév většinu hlavních složek ECM (tj. kolagen, elastin a proteoglykany) a také celou řadu enzymů, zodpovědných za udržení rovnováhy mezi syntézou a degradací ECM. V průběhu ATS však dochází ke stimulaci VSMCs působením LDL a atherogenních cytokinů, která způsobí změny ve složení ECM a následnou přestavbu cévní stěny. Atherogenní stimuly vyvolají změnu fenotypu VSMCs z kontraktilního na syntetický (aktivně proliferující buňky), který produkuje více ECM a má schopnost migrovat. Migrace VSMCs z medie do intimy je klíčová pro ztluštění intimy a přestavbu cévní stěny. VSMCs, které se dostaly do intimy, se díky expresi receptorů umožňujících příjem cholesterolu (např. LDL-R, LRP, VLDL-R, CD36) podílejí na hromadění lipidů v ATS plaku. VSMCs naplněné lipidy mají menší schopnost migrace a reparace, takže ATS plak je náchylnější k ruptuře. Zatímco buněčnou složku počáteční léze tvoří z 90-95 % VSMCs, v pozdních fázích tento podíl klesá na až 50 %.
Aktivace/adheze trombocytů a jejich agregace
Trombocyty za normálního stavu volně cirkulují v krvi v neaktivovaném stavu a neadherují k intaktnímu endotelu, k jejich aktivaci dochází během hemostázy nebo trombózy. Zánětlivé procesy probíhající v časných fázích ATS způsobují aktivaci endotelu a mohou tak stimulovat adhezi destiček. Zralý stabilní ATS plát obsahuje lipidové jádro kryté fibrózní čepičkou. Na celistvosti fibrózního krytu, který je tvořen kolagenními vlákny produkovanými VSMCs, závisí stabilita celého plátu. Mediátory zánětu a MMPs produkované makrofágy rozkládají fibrózní čepičku a narušují tak stabilitu ATS plátu. Při jeho roztrhnutí se lipidové jádro setká s krví, a protože je vnitřek plátu silně trombogenní (obsahuje tkáňový faktor, produkovaný makrofágy a pěnovými buňkami, a složky ECM). Na odkrytá kolagenní vlákna adherují prostřednictvím von Willebrandova faktoru trombocyty, čímž dochází k jejich aktivaci a také k aktivaci vnější cesty krevního srážení. Tkáňový faktor se váže na faktor VII/VIIa, který spolu s fosfolipidy a Ca2+ vytváří komplex, který aktivuje faktor X na Xa a podporuje tak lokální tvorbu trombinu. Ten vyvolává aktivaci destiček a na povrchu prasklého plátu se rychle tvoří trombus (Obr. 6). Faktor VIIa navíc aktivuje i faktor IX na IXa a tím propojuje vnitřní a vnější cestu hemokoagulace.
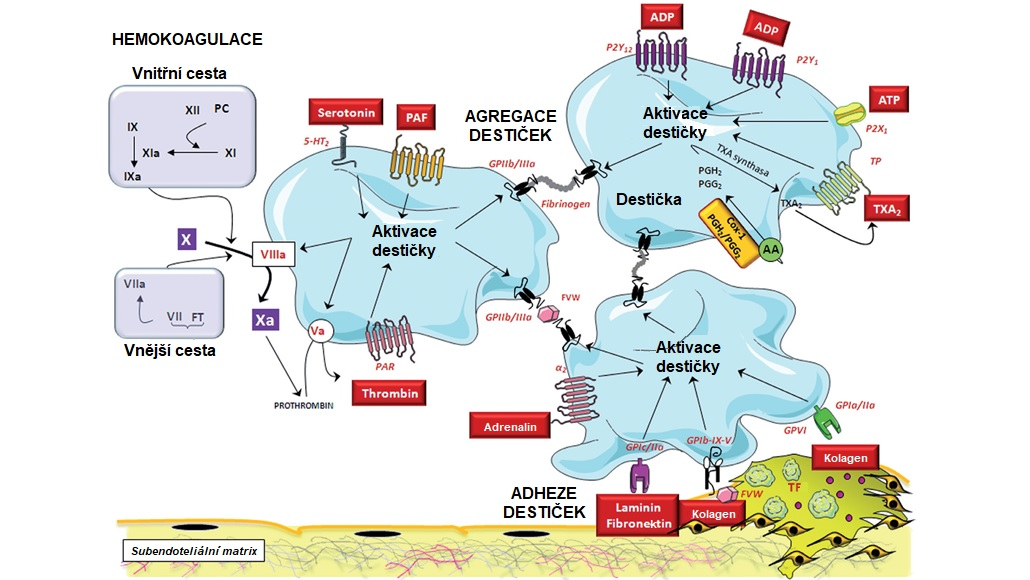
Obr. 6 Mechanismy účastnící se adheze, aktivace a agregace trombocytů (upraveno z Badimon et al. 2012)
AA, kyselina arachidonová; COX1, cyklooxygenasa 1; GP, glykoprotein; PAR, receptor aktivovaný proteasou; PC, protein C; PG, prostaglandin; TF, tkáňový faktor, TP, receptor pro tromboxan; VWF, von Willebrandův faktor
K aktivaci destiček dochází po interakci trombogenních substrátů s příslušnými receptory na jejich povrchu (Obr. 7). Kolagen aktivuje prostřednictvím svého receptoru na membráně trombocytů fosfolipasu A2 (aktivována rovněž Ca2+ a trombinem prostřednictvím R1), která uvolňuje arachidonát z fosfolipidů plasmatické membrány destičky. Z arachidonátu se syntetizuje tromboxan A2 (TxA2), který je z destičky uvolněn a prostřednictvím interakce s receptorem (R3) stimuluje fosfolipasu C, která rozkládá fosfatidylinositoldifosfát (PIP2) v plasmatické membráně trombocytu na diacylglycerol (DAG) a inositoltrifosfát (IP3). IP3 zprostředkovává vzestup Ca2+ v trombocytu. DAG a Ca2+ aktivují proteinkinasu C, která fosforyluje protein plekstrin a zahajuje uvolnění granul z aktivovaného trombocytu. Zvýšení koncentrací Ca2+ rovněž aktivuje fosforylaci lehkého řetězce myosinu, což vede ke změně tvaru trombocytu. Z destičkových granul je uvolněn adenosindifosfát (ADP), který má proagregační vlastnosti, a prostřednictvím aktivace svého receptoru (R4) a následné signalizační kaskády způsobí vystavení aktivovanějšího glykoproteinu IIb/IIIa (GPIIb/IIIa) na povrchu trombocytu. Tyto glykoproteiny slouží jako integrinové receptory, které váží dimery fibrinogenu a zprostředkovávají tak interakci mezi trombocyty a fibrinogenem a propojení sousedních trombocytů. Aktivované trombocyty se tak v místě léze shlukují a vytvářejí trombus. Antiagregační látkou, která deaktivuje aktivované trombocyty je PGI2 (tvořený buňkami endotelu), který aktivuje po vazbě na receptor (R5) adenylátcyklasu. Ta tvoří cAMP, který brání aktivaci trombocytu díky snížení intracelulární koncentrace Ca2+.
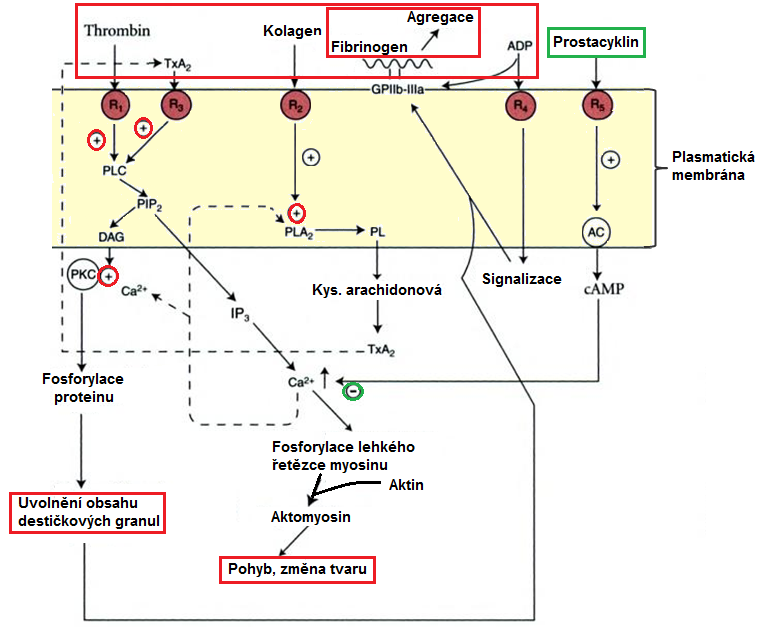
Obr. 7 Aktivace trombocytů (upraveno z Murray et al. 1990).
Nerovnováha mezi koagulací a fibrinolýzou vede k intravaskulární trombóze. Plasmin, součást fibrinolytického systému, rozpouští fibrinová koagula a tím udržuje plynulý průtok krve. Plasmin vzniká z plasminogenu syntetizovaného v játrech a uvolněného do krevního oběhu. Vznik plasminu je regulován tPA, jehož aktivita je regulována pomocí PAI-1 (Obr. 8). LDL i oxLDL zvyšují uvolnění PAI-1 a snižují uvolnění tPA z endotelu a tím snižují fibrinolytickou aktivitu. oxLDL navíc zvyšuje transkripci genu pro PAI-1 v endotelu a jeho uvolnění.
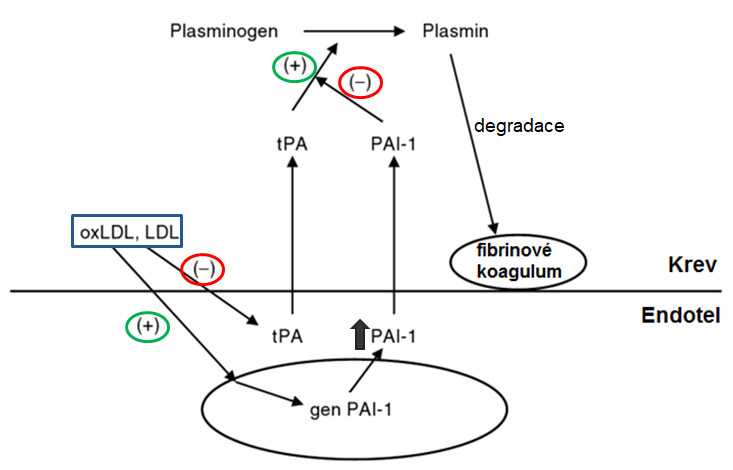
Obr. 8 Vliv LDL a oxLDL na fibrinolytický systém (upraveno z Kaur 2006)