Metabolismus sirných aminokyselin a jeho poruchy
Ze sirných aminokyselin je esenciální pouze methionin, cystein a cystin jsou v těle syntetizovány právě z methioninu. Metabolismus methioninu, homocysteinu a cysteinu jsou spojeny v cyklu methylace a transsulfurace (Obr. 10). V metabolismu methioninu bylo popsáno několik dědičných poruch, z nichž nejvýznamnější je homocystinurie, která může vzniknout díky deficitu cystathionin-β-synthasy nebo methylentetrahydrofolátreduktasy. S metabolismem cystinu jsou spojeny dvě transportní poruchy a to cystinurie a cystinosa.
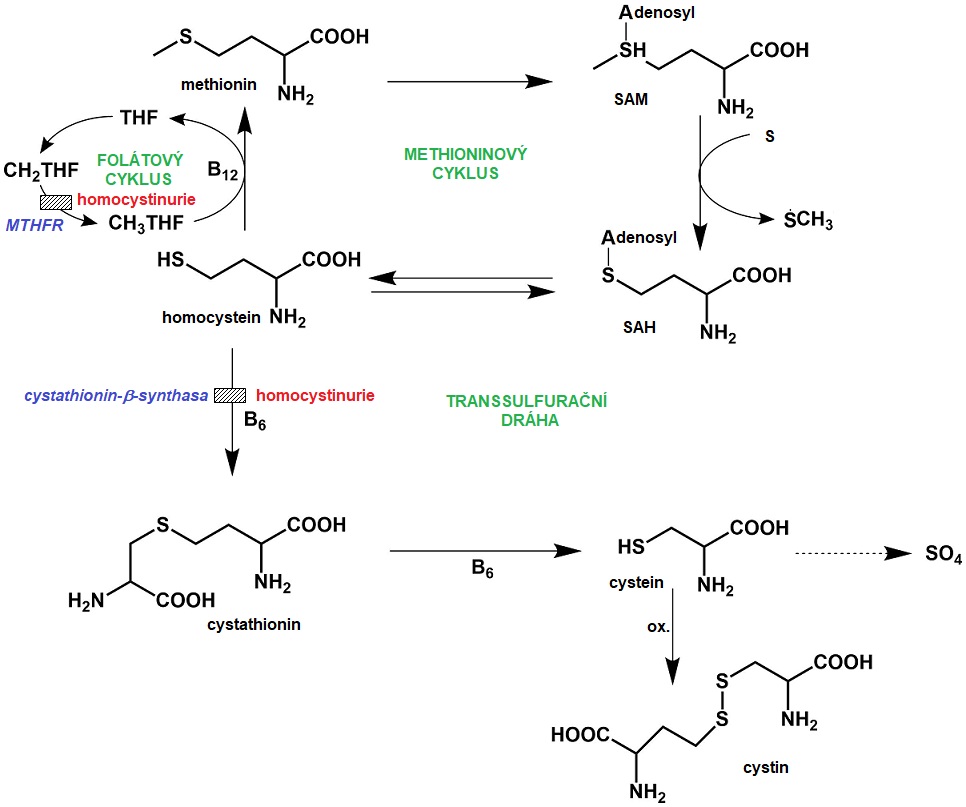
Obr. 10. Metabolismus sirných aminokyselin a poruchy metabolismu methioninu. SAM, S adenosylmethionin; SAH, S-adenosylhomocystein, THF, tetrahydrofolát; CH2THF, methylenTHF; CH3THF, methylTHF; MTHFR, methionintetrahydrofolátreduktasa
Homocystinurie
Homocystinurie může vzniknout z deficitu cystathionin-β-synthasy nebo z deficitu methylentetrahydrofolátreduktasy. V obou případech se zvyšují plasmatické koncentrace homocysteinu i jeho vylučování močí. Vysoké hladiny homocysteinu jsou nezávislým rizikovým faktorem žilní trombosy, byla rovněž nalezena souvislost s atherosklerosou.
Homocystinurie z deficitu cystathionin-β-synthasy (klasická homocystinurie) je autosomálně recesivně děděné onemocnění (incidence 1 : 6400 až 1 : 20500) a jedná se o vůbec nejčastější poruchu metabolismu aminokyselin obsahujících síru, která představuje blok v transsulfuraci. Při tomto deficitu vázne kondenzace homocysteinu a serinu na cystathionin. Koncentrace cystathioninu v organismu jsou snížené, zatímco koncentrace homocysteinu, jeho derivátů a methioninu, který vzniká remethylací homocysteinu, jsou zvýšené. Dosud bylo popsáno více než 160 mutací genu CBS, který kóduje cystathionin-β-synthasu. Tento enzym využívá jako kofaktor pyridoxal-5-fosfát, derivát vitaminu B6 (pyridoxinu).
Symptomy se objevují v ranném až pozdním dětství a zahrnují triádu postižení – pojivo, hemokoagulace a CNS. Starší neléčené děti mají mentální postižení, marfanoidní habitus (vysoká hubená postava, dlouhé tenké končetiny a prsty), dislokaci očních čoček a vysoké riziko trombosy a tromboembolických komplikací. Existují i lehčí formy onemocnění, u některých forem je průběh bezpříznakový. Důležitá je včasná diagnosa, která umožňuje normální vývoj i u pacientů s těžkou formou onemocnění. Proto je homocystinurie z deficitu cystathionin-β-synthasy zařazena do novorozeneckého screeningového programu. Je dostupná prenatální diagnostika na úrovni genu (je-li známa mutace v rodině) v choriových klcích i amniocytech, aktivita enzymu je měřitelná pouze v amniocytech. Léčba spočívá v omezení příjmu methioninu potravou s doplněním cysteinu, suplementaci cholinu či betainu (umožňují remethylaci homocysteinu), některé formy odpovídají na podávání pyridoxinu.
Homocystinurie z deficitu methylentetrahydrofolátreduktasy (MTHFR) je rovněž děděna autosomálně recesivně. MTHFR je nezbytná pro remethylaci homocysteinu na methionin, redukuje totiž 5,10-methylentetrahydrofolát na 5 methyltetrahydrofolát, který je zdrojem methylové skupiny. Díky tomu v organismu klesají hladiny methioninu a hromadí se homocystein, který je transsulfurační drahou přeměňován na cystathionin.
U těžkých infantilních forem se onemocnění projevuje progredujícím neurodegenerativním postižením (poškození bílé hmoty mozkové; psychomotorická retardace již v prvních dvou letech života) s hypotonií, opakovanými atakami apnoe, mikrocefalií a tromboembolickými poruchami. U lehčích forem onemocnění a heterozygotů je zvýšené kardiovaskulární riziko díky protrombofilnímu stavu. U neléčených pacientů je infantilní forma smrtelná, zatímco při včasné diagnose je možné léčbou zastavit psychomotorické postižení. To je pravděpodobně důvod, proč bylo toto onemocnění zařazeno do novorozeneckého screeningového programu. Při prenatální diagnostice se provádí vyšetření enzymové aktivity a/nebo vyšetření familiárních mutací v genu MTHFR. Léčba je založena na podávání vysokých dávek betainu, podle individuální odezvy je možné doplnit methionin, kyselinu listovou, 5-methylentetrahydrofolát a vitaminy B6 a B12 v různých kombinacích.
Cystinurie
Cystinurie (incidence 1 : 7000) je transportní porucha způsobená defektem vysokoafinitního luminálního přenašeče pro cystin a dibazické aminokyseliny v buňkách proximálních tubulů ledvin a mukosy jejuna. V jejím důsledku je snížená absorpce cystinu v tenkém střevě i jeho reabsorpce v ledvinách, cystin je ve vysokých koncentracích vylučován močí. Vzhledem k velmi malé rozpustnosti cystinu v neutrálním či slabě kyselém pH moči vznikají v ledvinách cystinové konkrementy a pacient je celoživotně ohrožen urolithiasou. Cystinový transportér je složený ze dvou různých podjednotek, které jsou kódovány geny SLC3A1 a SLC7A9 (SLC, „solute carrier"). Současná klasifikace je založena na nalezené mutaci – u typu A (dědičnost autosomálně recesivní) je mutován gen SLC3A1, u typu B (dědičnost autosomálně dominantní s neúplnou penetrancí) je mutován gen SLC7A9, u vzácného typu AB jsou mutované obě allely jednoho genu a navíc jedna alela druhého genu (tedy AAB nebo ABB).
Cystinurie je charakterizována vznikem ledvinových kamenů, které se obvykle objevují ve druhé dekádě života. U některých pacientů se mohou první kameny objevit již v ranném dětství, jiní mohou být celoživotně bez potíží. Urolithiasa se téměř vždy objevuje u typu A, u ostatních dvou typů se vyskytuje nekonstantně. Kameny jsou extrémně tvrdé a žlutohnědé, krystaly cystinu v moči mají šestiúhelníkový tvar. Diagnosa je založena na laboratorní analýze moči – nález hexagonálních krystalů, pozitivní Brandova zkouška (NaCN konvertuje cystin na cystein a po přidání nitroprussidu sodného vzniká rudé zbarvení moči) a kvantitativní analýza aminokyselin v moči/24 hodin. Základem léčby je pitný režim (3-4 litry tekutin/den) a alkalizace moči, která zvyšuje rozpustnost cystinu. Rozpustnost cystinu lze rovněž zvýšit podáním chelátorů (penicilamin, bucilamin, thiopronin). V některých případech může být nezbytné chirurgické odstranění kamenů.
Cystinosa
Cystinosa (incidence 1 : 180 000) je autosomálně recesivně děděný deficit lyzosomálního transportéru pro cystin, cystinosinu. Při defektu cystinosinu se v lyzozomech buněk celého organismu hromadí cystin. Nejvíce poškozeny jsou ledviny, cystinosa je také nejčastější příčinou Fanconiho syndromu. Podle klinického průběhu rozlišujeme tři formy onemocnění: nejzávažnější infantilní formu, juvenilní formu a okulární formu. Prvními klinickými znaky infantilní cystinosy je Fanconiho syndrom s polyurií a polydypsií před dosažením 1. roku věku. Poškození renálních funkcí přechází do chronického selhávání ledvin. Běžné jsou poruchy růstu (trpaslictví) a vitamin D-rezistentní rachitis, do tří let věku se rozvíjí fotofobie a poškození zraku. Mezi pozdní komplikace patří hypothyreosa, hepatosplenomegalie, ulcerace rohovky, respirační insuficience a dysfagie. Bez léčby dochází na konci první dekády života k fatálnímu selhání ledvin. U juvenilní formy je v popředí postižení ledvin, zejména glomerulů s proteinurií, neúplným Fanconiho syndromem a postupným selháváním ledvin. Okulární (též benigní) forma je charakterizována retinopatií, protože cystinové krystaly se ukládají v rohovce oka, která se stává neprůhlednou.
Diagnosa je stanovena na základě biochemického průkazu zvýšených intracelulárních hladin cystinu v leukocytech. Přítomnost cystinových krystalů v rohovce lze zjistit pomocí štěrbinové lampy. Léčba se v současnosti opírá o podávání cysteaminu (p.o., oční kapky), sloučeniny, která v lyzosomech štěpí cystin na dva cysteiny a s jednou molekulou cysteinu vytváří smíšený disulfid, který je z lyzosomu přenášen transportérem pro lysin. Druhý cystein může být z lyzosomů přenesen do cytoplasmy pomocí transportéru pro cystein. Cysteamin oddaluje selhání ledvin, snižuje poškození štítné žlázy a očí a zlepšuje růst. Jeho nevýhodou je, že nevede k úpravě Fanconiho syndromu, jeho použití je provázeno nepříjemným zápachem dechu a gastrointestinálními potížemi. Používá se rovněž podpůrná léčba tubulárních ztrát (např. hydratace, ionty, vit. D, karnitin), podávání růstového hormonu a thyroxinu.