Metabolismus větvených aminokyselin a jeho poruchy
Katabolismus tří esenciálních větvených aminokyselin, leucinu, isoleucinu a valinu, probíhá zpočátku společnou metabolickou drahou (Obr. 8). Tyto aminokyseliny jsou nejprve transaminovány na odpovídající 2-oxokyseliny aminotransferasou větvených aminokyselin, která má vysokou aktivitu zejména ve svalech a myokardu a nízkou v játrech. Vznikající 2 oxokyseliny následně podstupují oxidační dekarboxylaci na acyl-CoA dehydrogenasou větvených 2-oxokyselin (BCKD). Poté se degradační dráha rozděluje. Leucin je degradován na acetoacetát a acetyl-CoA, který vstupuje do citrátového cyklu. Isoleucin je štěpen na acetyl CoA a propionyl-CoA, který vstupuje do citrátového cyklu po přeměně na sukcinyl CoA. Valin je rovněž metabolizován na propionyl-CoA. V důsledku deficitu specifických enzymů katabolismu větvených aminokyselin vzniká skupina poruch označovaných jako organické acidurie (např. leucinosa, isovalerová acidurie, propionová acidurie, methylmalonová acidurie; tato čtyři onemocnění mají celou řadu společných klinických i biochemických symptomů).
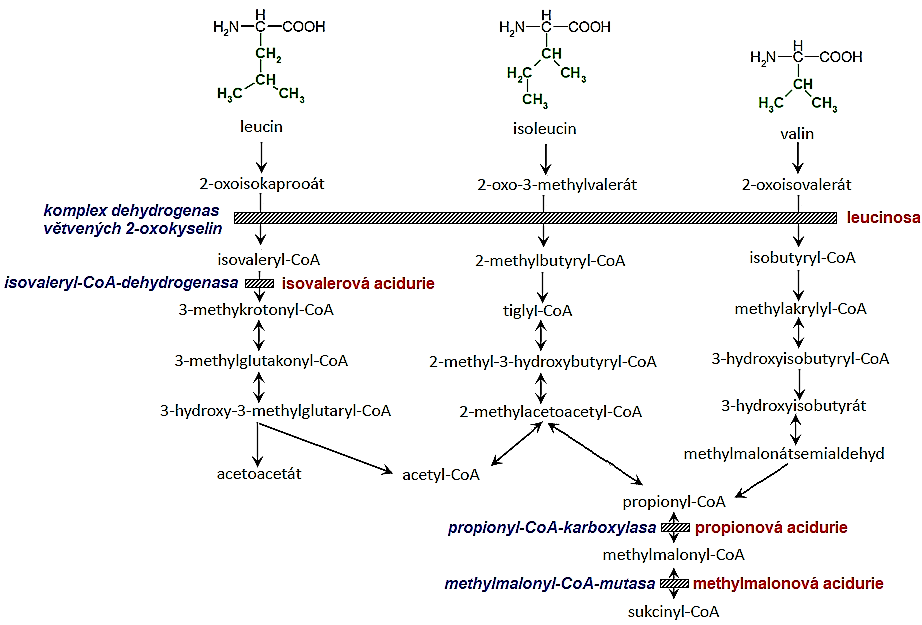
Obr. 8. Katabolismus větvených aminokyselin a jeho poruchy.
Leucinosa
Leucinosa, též označovaná jako choroba javorového sirupu („maple syrup urine disease", MSUD), je autosomálně recesivní onemocnění (incidence 1 : 185 000) způsobené deficitem komplexu dehydrogenas větvených 2-oxokyselin (BCKD). BCKD je multienzymový komplex (podobá se pyruvátdehydrogenasovému komplexu) volně asociovaný s vnitřní mitochondriální membránou a se skládá ze tří katalytických součástí (dekarboxylasa složená ze dvou podjednotek, dihydrolipoylacyltransferasa, dihydrolipoamiddehydrogenasa) a 2 regulačních enzymů. Leucinosu vyvolávají defekty v podjednotkách dekarboxylasy (využívá jako kofaktor thiamin) nebo v dihydrolipoylacyltransferase, defekt v posledním enzymu způsobuje specifický syndrom s laktátovou acidosou. Při leucinose se v krvi, moči a likvoru výrazně zvyšuje koncentrace větvených 2-oxokyselin a také větvených aminokyselin, protože úvodní transaminační reakce je vratná. V krvi pacientů s leucinosou se nachází také stereoisomer isoleucinu, alloisoleucin. Tato látka je nejspecifičtějším a nejcitlivějším diagnostickým markerem všech forem leucinosy. Ačkoliv se postihuje defekt BCDK metabolismus všech větvených aminokyselin, neurotoxickými formami jsou leucin a jeho 2-oxokyselina, kyselina 2-oxoisokaproová, proto je onemocnění označované jako leucinosa. Tyto látky jsou v plasmě přítomné v ekvimolárním množství, a pokud jejich hladiny přesáhnou koncentraci 1 mM, mohou způsobit akutní mozkovou dysfunkci. MSUD je součástí novorozeneckého screeningového programu a včasné odhalení této choroby může zlepšit prognózu pacientů.
Leucinosa se projevuje již koncem prvního týdne života a neléčená je smrtelná. Vyskytuje se několik forem: klasická těžká MSUD, intermitentní MSUD, mírná MSUD a thiamin-responzivní MSUD. Typickým znakem všech forem je nasládlý zápach moči po javorovém sirupu. U klasické těžké MSUD dojde u donošených novorozenců (po normálním těhotenství i porodu) po bezpříznakovém období náhle ke zhoršení stavu (potíže s krmením, spavost, progresivní kóma), které neodpovídá na symptomatickou léčbu. V komatózním stavu se často objevují myoklonické záškuby. Pokud dítě přežije akutní fázi, je stále ohroženo ketoacidosou, akutním edémem mozku a exitem po zátěži (např. infekcí, operačním výkonem). Při špatné kompenzaci a trvalém zvýšení hladin větvených aminokyselin se rozvíjí psychomotorická retardace (dochází k demyelinizaci nervových vláken). Mírnější formy MSUD se objevují později v kojeneckém a batolecím věku a neléčené jsou provázeny psychomotorickou retardací.
Léčba je symptomatická a je založená na omezení příjmu valinu, leucinu a isoleucinu. V akutní fázi je u novorozenců s těžkou MSUD potřeba komplexní léčba spolu s neprodleným odstraněním toxinů (např. peritoneální dialýzou). Dieta však neovlivní již vzniklé neurologické a mentální postižení. Pacienti s těžkou MSUD mají obvykle výrazně nižší IQ než srovnatelná populace a dosažený intelekt je nepřímo úměrný době, kdy zůstávaly po narození hladiny leucinu v plasmě nad 1 mM. Některé intermitentní a mírné formy MSUD dobře reagují na podávání thiaminu.
Isovalerová acidurie
Jedná se o autosomálně recesivní onemocnění (incidence 1 : 230 000) vyvolané deficitem isovaleryl CoA dehydrogenasy. Tento defekt vede k hromadění isovaleryl-CoA a jeho metabolitů - volné kyseliny isovalerové, 3 hydroxyisovalerové kyseliny, N-isovalerylglycinu a isovalerylkarnitinu. Isovalerát způsobuje typický zápach zpocených nohou. Děti s akutní neonatální formou mají po několika dnech normálního vývoje problémy s krmením, zvracení, těžkou metabolickou ketoacidosu a bez léčby progredují ke kómatu a úmrtí. Často je přítomna dehydratace, hyperamonemie, trombocytopenie a neutropenie. Zhruba polovina pacientů s akutní formou choroby umírá při první atace. Ti, kteří přežijí, mají neurologické poškození, které se však může zcela upravit. Chronická intermitentní forma se prezentuje v průběhu dětství epizodami metabolické acidosy obvykle při metabolickém stresu (např. infekce, zvýšený příjem proteinů). Pacienti s touto formou onemocnění mohou mít neurologické postižení, ale obvykle je růst a vývoj normální. K úmrtí při acidotické atace však může dojít v kterémkoliv věku.
Isovalerová acidurie je součástí novorozeneckého screeningového programu. Prenatální diagnostika se provádí analýzou isovalerylglycinu v amniové tekutině a/nebo enzymatickou nebo DNA analýzou v choriových klcích/amniocytech. Léčba sestává z nízkoproteinové diety s omezením leucinu spolu se suplementací glycinem a karnitinem. Glycin a karnitin umožňují netoxické odbourání nadbytečného isovaleryl-CoA.