Metabolismus aromatických aminokyselin a jeho poruchy
Katabolismus fenylalaninu a tyrosinu je dobré znát, protože poruchy degradace těchto aminokyselin jsou v této oblasti světa poměrně časté a klinicky závažné (Obr. 3). Katabolismus těchto aminokyselin nelze od sebe oddělovat. Kromě zmíněné degradační dráhy je menší množství fenylalaninu a tyrosinu metabolizováno dalšími drahami na biologicky významné produkty (např. pigment melanin, neurotransmitery dopamin a noradrenalin, hormony adrenalin, thyroxin a trijodtyronin).
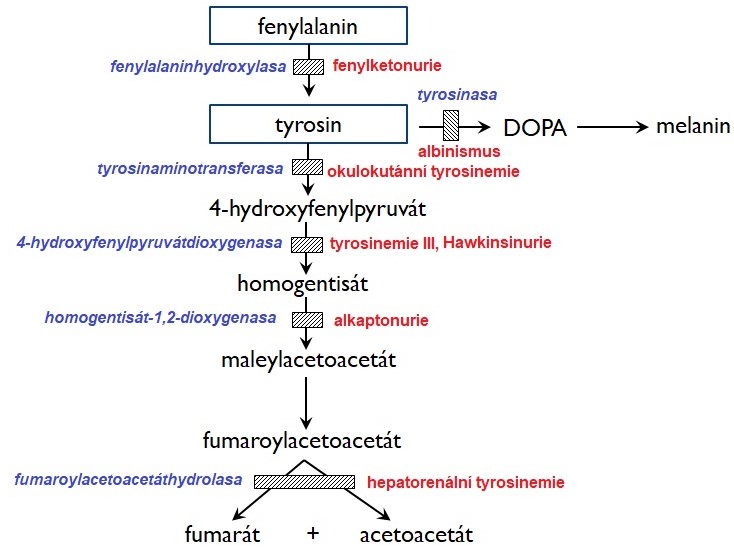
Obr. 3. Katabolismus fenylalaninu a tyrosinu a jeho poruchy
Fenylalanin je esenciální aromatická aminokyselina, jejíž metabolismus je v organismu zahájen ireversibilní hydroxylací na tyrosin, která je katalyzována fenylalaninhydroxylasou (PAH) (Obr. 4). Tento enzym využívá koenzym tetrahydrobiopterin (BH4), který je v organismu syntetizován třemi kroky z GTP. Během hydroxylace fenylalaninu je přeměněn na pterin-4a-karbinolamin, který je následně regenerován. Deficit PAH nebo poruchy v tvorbě či regeneraci BH4 způsobují hyperfenylalaninemii a deficit tyrosinu. Těžký deficit PAH je příčinou vzniku závažného onemocnění fenylketonurie (PKU), která neléčená způsobuje trvalé postižení CNS.
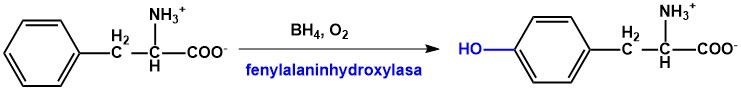
Obr. 4. Hydroxylace fenylalaninu na tyrosin katalyzovaná fenylalaninhydroxylasou
Tyrosin patří mezi nejméně rozpustné aminokyseliny. Zdrojem tyrosinu je jednak potrava, jednak hydroxylace fenylalaninu. Tyrosin je metabolizován na fumarát a acetoacetát. Vzdálené metabolity katabolismu tyrosinu, maleylacetoacetát a fumaroylacetoacetát, mohou být redukovány na sukcinylacetoacetát a následně dekarboxylovány za vzniku sukcinylacetonu, který je nejsilnějším inhibitorem ALA-dehydratasy, enzymu syntézy hemu. Je známo pět dědičných poruch katabolismu tyrosinu a albinismus.
Fenylketonurie
Fenylketonurie je dědičné metabolické onemocnění způsobené deficitem fenylalaninhydroxylasy. Jedná se o klinicky nejzávažnější poruchu metabolismu aromatických aminokyselin, která má v naší populaci poměrně vysokou frekvenci výskytu (1 : 6500). Dědičnost je autosomálně recesivní a onemocnění je provázeno hromaděním fenylalaninu a alternativních produktů jeho degradace. Fenylketonurie je klasifikována podle hladin fenylalaninu a zbytkové aktivity PAH na klasickou PKU, mírnou PKU a mírnou hyperfenylalaninemii (HPA). U klasické PKU jsou hladiny fenylalaninu v krvi při normální stravě vyšší než 1200 µM při normální stravě a zbytková aktivita PAH je menší než 1 %. U mírné PKU se hladiny fenylalaninu pohybují v rozmezí 600 1200 µM a zbytková aktivita PAH je 1-5 %. Koncentrace fenylalaninu v krvi u mírné HPA se pohybují v rozmezí 120-600 µM a zbytková aktivita PAH je vyšší než 5 %. Toto rozdělení je důležité kvůli indikaci dietní léčby. Kromě deficitu PAH se hyperfenylalaninemie objevuje u lidí s deficitem některého enzymu syntézy nebo recyklace tetrahydrobiopterinu, které byly dříve označovány jako maligní PKU nebo maligní HPA. Maligní PKU/HPA se léčí jiným způsobem než fenylketonurie.
PKU byla poprvé popsána v roce 1934 norským lékařem Ivarem Asbjørnem Føllingem jako „Imbecillitas phenylpyruvica". PKU se projeví až po porodu, kdy začnou hladiny fenylalaninu postupně stoupat, protože již nejsou odstraňovány placentou. Během kojeneckého a dětského věku dochází k progresivnímu ireverzibilnímu poškození mozku, u neléčených pacientů se rozvine mentální postižení, poruchy chování, neurologické a somatické poškození. Nejčastějšími projevy jsou těžká mentální retardace (IQ ≤ 50), zápach připomínající myšinu (díky vylučování alternativních produktů), hypopigmentace vlasů, kůže a očí (nedostatek pigmentu melaninu), křeče, kožní vyrážky (raš, ekzémy) a poruchy chování (např. hyperaktivita, agresivita, stereotypie, anxieta, sociální izolace). Klinický fenotyp dobře koreluje s hladinami fenylalaninu v krvi, které odpovídají stupni deficitu PAH.
Deficit PAH způsobí, že je fenylalanin metabolizován alternativní drahou, která je za normálních okolností nevýznamná (Obr. 5). Dochází k transaminaci fenylalaninu na fenylpyruvát, který se pak vylučuje močí – odtud pochází název tohoto onemocnění. Kromě fenylpyruvátu vznikají touto cestou i další vedlejší produkty, které se vylučují močí (např. fenylacetát, fenyllaktát). Fenylalanin může podstupovat dekarboxylaci na fenylethylamin nebo acetylaci na N acetylfenylalanin, které jsou rovněž přítomné ve vysoké koncentraci v moči pacientů s PKU. Vznikající fenylketony jsou příčinou zápachu po myšině, který je pro PKU typický.
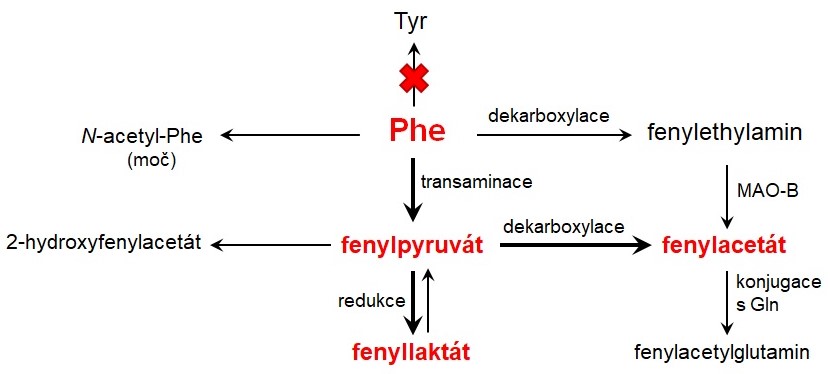
Obr. 5. Vznik alternativních produktů u fenylketonurie. Phe, fenylalanin; Tyr, tyrosin; Gln, glutamin; MAO-B, monoaminoxidasa-B
Patogeneze poškození mozku u PKU není ještě zcela objasněna, ale kauzálně souvisí se zvýšenými hladinami fenylalaninu v krvi. Vysoké hladiny Phe kompetitivně brání přenosu dalších dlouhých neutrálních aminokyselin (Trp, Tyr, Val, Leu, Ile, His, Met a Thr) do mozku díky saturaci transportéru LAT1 („L-Type Amino Acid Transporter"). V důsledku toho klesají v mozku hladiny těchto aminokyselin a bude tedy omezena syntéza neurotransmiterů dopaminu, noradrenalinu a serotoninu. Fenylalanin navíc působí jako inhibitor tyrosinhydroxylasy a tryptofanhydroxylasy, které se účastní syntézy těchto neurotransmiterů. Nedostatek esenciálních aminokyselin (např. Trp, u PKU i Tyr) způsobí omezení syntézy proteinů v mozku, která je nezbytná pro správný vývoj a fungování mozku. Nejznámějším specifickým příkladem je myelin, jehož struktura je u pacientů s PKU abnormální. Jeho součástí je bazický protein myelinu, jehož syntéza je narušena, i pokud chybí jen jedna esenciální aminokyselina. Rovněž syntéza dalších proteinů včetně enzymů (např. pyruvátkinasa, HMG-CoA-reduktasa, tyrosin- a tryptofanhydroxylasa) a receptorů je narušena při nízkých koncentracích esenciálních aminokyselin, což může vést ke snížení synaptické plasticity a růstu axonů. Vysoké koncentrace fenylalaninu inhibují aktivitu HMG CoA reduktasy, což se projeví sníženou syntézou cholesterolu, který je důležitou součástí myelinu. Fenylalanin pravděpodobně intrferuje s glutamátergní transmisí, synaptogenezí a aktivitou pyruvátkinasy, což vede k útlumu energetického metabolismu, který byl pozorován u pacientů s PKU.
PKU je detekována novorozeneckým screeningem, který je prováděn v suché kapce krve pomocí tandemové hmotnostní spektrometrie. U PKU jsou nalezeny vysoké hladiny fenylalaninu a abnormální poměr fenylalanin/tyrosin. Diagnosa je stanovena analýzou fenylalaninu a tyrosinu a vyloučením defektu pterinů /stanovení pterinů v moči a aktivity enzymu dihydropteridinreduktasy). Potvrzení diagnosy je provedeno DNA mutační analýzou. Je dostupná prenatální diagnostika, která se provádí DNA analýzou genu PAH v kultivovaných choriových klcích nebo amniocytech.
PKU je zatím nevyléčitelná, ale jejím následkům lze bránit dietou s nízkým obsahem fenylalaninu a podáváním směsi aminokyselin bez fenylalaninu. Dieta je doporučována po celý život a její dodržování je považováno za nejdůležitější faktor normálního vývoje mozku. Dostupné jsou kojenecké formule s aminokyselinami bez fenylalaninu. Sladidlo aspartam, které ve své molekule obsahuje fenylalanin, je pro dietu pacientů s PKU nevhodné. Mezi nové postupy patří podávání syntetického derivátu BH4 sapropterinu, dlouhých neutrálních aminokyselin, které kompetitivně blokují transport fenylalaninu do mozku, a glykomakropeptidu, což je jediný známý protein v potravě bez obsahu fenylalaninu. Ve fázi klinického zkoušení je ERT s rekombinantní fenylalaninamoniaklyasou modifikovanou polyethylenglykolem, která rozkládá fenylalanin na amoniak a kyselinu trans-skořicovou. Tento přípravek byl v roce 2018 v USA schválen FDA k užití u dospělých pacientů s PKU s hladinami fenylalaninu přesahujícími 600 µM. Genová terapie je ve stádiu výzkumu.
U matek s PKU musí být během těhotenství hladiny fenylalaninu důkladně monitorovány a udržovány v bezpečných mezích (120-240 µM). Vysoké koncentrace fenylalaninu během těhotenství jsou totiž spojeny s rozvojem specifického syndromu, tzv. maternální PKU, který se projevuje faciální dysmorfií, mikrocefalií, psychomotorickou retardací, poruchami učení a vrozenými srdečními vadami. Důvodem je, že koncentrace fenylalaninu jsou v plodu 1,5-2x vyšší než hladiny u matky kvůli aktivnímu transportu látek z matky do plodu. Fenylalanin kompetitivně ovlivňuje transport dalších dlouhých neutrálních aminokyselin a ovlivňuje tak vývoj plodu. Ženy s PKU by měly již 3-6 měsíců před otěhotněním dodržovat nízkobílkovinnou dietu a snažit se o početí až ve chvíli, kdy jsou hladiny fenylalaninu v krvi stabilní a nižší než 240 µM.
Albinismus
Melanin, tmavý pigment zodpovědný za zbarvení kůže, vlasů a očí, je syntetizován z tyrosinu v melanosomech melanocytů. Syntézu zahajuje enzym tyrosinasa (Obr. 6), která je specifická pro melanocyty. Pokud je syntéza, transport či distribuce melaninu defektní, vzniká albinismus. Postižení jedinci mají světlou pleť a vlasy a průsvitné modré oční duhovky, což způsobuje snížení zrakové ostrosti, nystagmus, strabismus a fotofobii, případně až slepotu. Kůže albínů je citlivá ke slunečnímu záření a náchylná ke vzniku karcinomů kůže.
Onemocnění se vyskytuje v několika základních variantách: okulokutánní (generalizovaná forma postihující vlasy, kůži a oči), okulární (postihuje pouze oči) a parciální (lokální hypopigmentace kůže a vlasů, nepostihuje oči). U okulokutánního albinismu byly identifikovány čtyři autosomálně recesivně děděné geny, které jsou zodpovědné za vznik tohoto onemocnění. Jedná se o enzym tyrosinasu a tři proteiny zajišťující transport a distribuci melaninu. Okulokutánní albinismus se vyskytuje ve dvou formách – tyrosinasa-pozitivní a tyrosinasa-negativní. Nejčastější formou albinismu je tyrosinasa-negativní typ, který je způsoben deficitem tyrosinasy v melanocytech. Postižení jedinci nejsou schopni syntetizovat melanin a mají typické projevy na kůži, vlasech a očních duhovkách. Tyrosinasa-pozitivní typ je druhou nejčastější formou albinismu, onemocnění je způsobeno defektem v distribuci melaninu. U jedinců s okulárním albinismem je postižení omezeno na oční duhovky. Toto onemocnění má dědičnost vázanou na chromosom X a závažnější formy jsou často provázeny hluchotou.
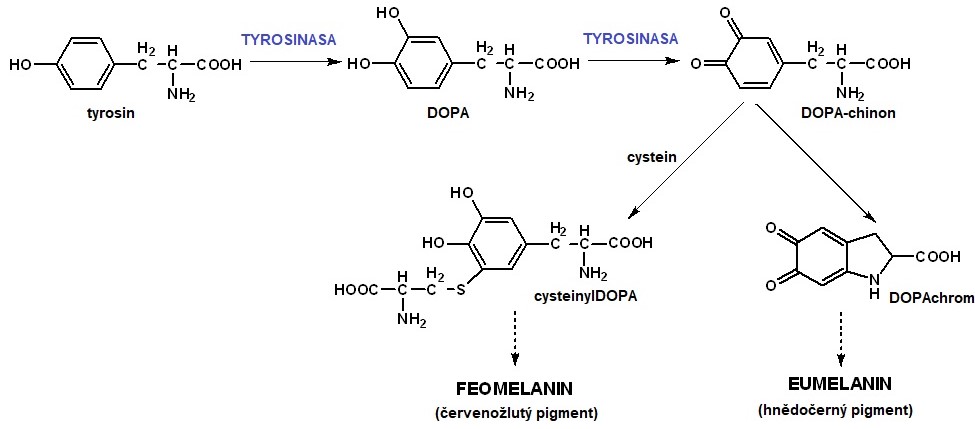
Obr. 6. Syntéza melaninů
Alkaptonurie
Alkaptonurie je způsobena deficitem homogentisát-1,2-dioxygenasy, enzymu štěpícího homogentisát na maleylacetoacetát. Tento enzym je exprimován hlavně v játrech a ledvinách. Homogentisát se ve velkém množství vylučuje močí, která na vzduchu vlivem oxidace tmavne díky tvorbě tmavého pigmentu alkaptonu, který je podobný melaninu. Alkapton a jeho prekurzor benzochinonacetát se ukládají v různých tkáních (zejm. chrupavky, skléry a kosti) a způsobují jejich tmavé zbarvení – ochronosu (řec. ochros - žlutohnědý, nosos – nemoc).
Onemocnění je autosomálně recesivní s frekvencí výskytu 1 : 100-250 000. Zajímavostí je, že nejvyšší prevalenci má alkaptonurie na Slovensku (1 : 19 000). Patří mezi méně závažná metabolická onemocnění, protože nepostihuje nervový systém. Klinické příznaky onemocnění se začínají objevovat až v dospělosti (3. – 4. dekáda) postižením kloubů podobné osteoartritidě, postižením srdce a urolithiasou. Ochronosa vede k zánětlivým a degenerativním změnám kloubů s omezením hybnosti a bolestí.
Tyrosinemie
Tyrosinemie je označení vrozených poruch katabolismu tyrosinu, v jejichž důsledku dochází ke zvýšení hladin tyrosinu v krvi nad 200 µM. Dosud byla popsána čtyři onemocnění: tyrosinemie typu I (hepatorenální), tyrosinemie typu II (okulokutánní), tyrosinemie typu III a Hawkinsurie. Kromě Hawkinsurie, která je děděna autosomálně dominantně, jsou ostatní tři onemocnění děděna autosomálně recesivně.
Tyrosinemie typu I je způsobena deficitem fumaroylacetoacetáthydrolasy, která je exprimovaná zejména v játrech a ledvinách. V důsledku deficitu tohoto enzymu dochází k hromadění metabolitů předcházejících tento blok, maleylacetoacetátu a fumaroylacetátu, a jejich derivátů sukcinylacetonu a sukcinylacetoacetátu (Obr. 7). Tyto látky mají významné patogenní účinky. Sukcinylaceton působí jako inhibitor 4 hydroxyfenylpyruvátdioxygenasy, což vede k vzestupu plasmatických hladin tyrosinu. Rovněž inhibuje ALA-dehydratasy v játrech a erytrocytech, což způsobí útlum syntézy hemu, zvýšení hladin δ-aminolevulové kyseliny (ALA) a její zvýšenou exkreci močí. Zvýšené hladiny δ-ALA jsou příčinou akutních neurologických krizí („porfyrická krize"). Hromadění fumaroylacetoacetátu a maleylacetoacetátu v hepatocytech a buňkách renálních tubulů vyvolává jejich apoptosu nebo významnou alteraci genové exprese (působí jako alkylační činidla). Tyto metabolity narušují metabolismus thiolů (např. glutathionu) a tak činí buňky náchylnější k oxidačnímu stresu.
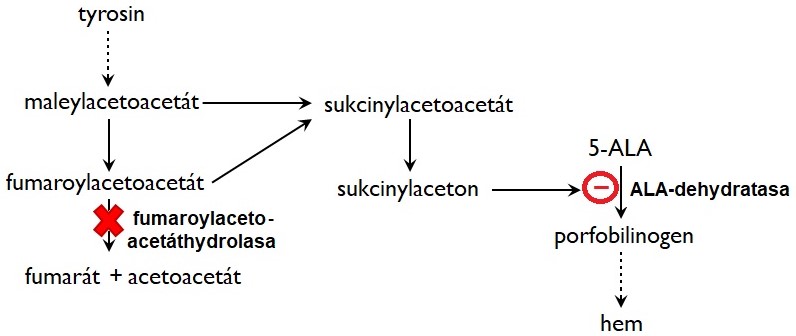
Obr. 7. Metabolické odchylky u hepatorenální tyrosinemie
Klinicky se tyrosinemie typu I klasifikuje na základě věku, kdy se objevují symptomy, na akutní, subakutní a chronickou formu. Akutní forma se manifestuje do 6 měsíců života dítěte opožděným psychomotorickým vývojem, zvracením, průjmy a následně příznaky akutního selhání jater. Děti umírají na jaterní selhání do 2 let života. Chronická forma se manifestuje po prvním roce života neprospíváním, progresivní cirhosou, Fanconiho syndromem s hypofosfatemickou křivicí a porfyrickými krizemi. Je zde vysoké riziko vzniku hepatocelulárního karcinomu. Děti umírají kolem desátého roku života.
V minulosti bylo toto onemocnění léčeno dietou s nízkým obsahem fenylalaninu a tyrosinu, s nebo bez transplantace jater. V současnosti je transplantace jater omezena na pacienty s akutním selháním jater, kteří nereagují na léčbu nitisinonem, a na pacienty s podezřením na hepatocelulární karcinom. V terapii se nyní používá zejména nitisinon, což je silný inhibitor 4-hydroxyfenylpyruvátdioxygenasy. Dochází tedy k zastavení katabolismu tyrosinu v časném kroku, takže nevznikají toxické metabolity, současně však stoupají hladiny tyrosinu a 4-hydroxyfenylpyruvátu. Je tedy nutné omezit příjem tyrosinu a fenylalaninu dietou, aby se zabránilo nežádoucím účinkům hypertyrosinemie. Malá část pacientů s akutními projevy na léčbu nitisinonem nereaguje, u nich postižení jater dále progreduje a bez urgentní transplantace ledvin je mortalita velmi vysoká.
Tyrosinemie typu II je vyvolána deficitem jaterní tyrosinaminotransferasy, což vede k výraznému vzestupu koncentrace tyrosinu v krvi (> 1200 µM) a likvoru. Onemocnění se projevuje očními lézemi (fotofobie, slzení, bolestivé pálení očí, herpetiformní ulcerace), kožními lézemi (puchýře a bolestivé eroze s krustami na dlaních a ploskách) a neurologickými komplikacemi (variabilní, různý stupeň opoždění vývoje). Příčinou vzniku kožních lézí je pravděpodobně krystalizace tyrosinu v epiteliálních buňkách rohovky, která vyvolává zánětlivou odpověď. Léčba sestává z diety s omezením fenylalaninu a tyrosinu.
Tyrosinemie typu III a Hawkinsinurie jsou velmi vzácná onemocnění s malým počtem klinicky popsaných případů a obě jsou vyvolaná deficitem 4-hydroxyfenylpyruvátdioxygenasy. Tyrosinemie typu III může probíhat asymptomaticky nebo je spojená s mentální retardací a je provázena zvýšenými hladinami tyrosinu. Hawkinsinurie může být asymptomatická nebo se projevuje v dětském věku neprospíváním a metabolickou acidosou. U této choroby byly popsány abnormální metabolity (např. hawkinsin), které vznikají při nekompletní přeměně 4-hydroxyfenylpyruvátu na homogentisát.