Intracelulární degradace proteinů
Tělesné proteiny se neustále obměňují. Zatímco některé proteiny jsou velmi stabilní s dlouhým poločasem rozpadu, jiné mají poločas rozpadu jen v řádu minut. Velmi rychle jsou degradovány regulatorní proteiny (např. transkripční faktory). Rozklad těchto proteinů umožní aktivaci, nebo naopak zastavení určité signální dráhy. Rovněž proteiny s abnormálním složením (např. mutantní hemoglobiny, výsledek chyb v translaci či skládání, poškozené oxidací či jinou cestou) musí být rychle eliminovány, protože hrozí jejich nahromadění v buňce a tvorba agregátů, které mohou buňku poškodit či zahubit. Posledním důvodem degradace intracelulárních proteinů je metabolická potřeba, kdy jsou uvolněné aminokyseliny využity jako zdroj energie. Degradace tkáňových proteinů musí být citlivě regulována.
Intracelulární degradace proteinů probíhá buď v lyzosomech, nebo v cytosolu. Extracelulární, membránové a proteiny s dlouhým biologickým poločasem jsou degradovány v lyzosomech drahou nezávislou na ATP, zatímco rozklad regulatorních a poškozených proteinů probíhá v cytosolu drahou, která je závislá na přísunu ATP a na přítomnosti specifického proteinu – ubikvitinu (UBQ). Kromě těchto dvou nejvýznamnějších drah mohou být intracelulární proteiny štěpeny i pomocí kalpainů, což jsou cytolové ATP-dependentní cysteinové peptidasy závislé na Ca2+, které se podílejí na regulaci intracelulárních procesů spojených se změnami v koncentraci Ca2+ (např. regulace buněčného cyklu, proteinkinasy C, transkripčních faktorů, přenosu signálu a apoptosy). Posledním typem intracelulární proteolýzy je apoptosa zprostředkovaná cysteinovými peptidasami kaspasami (viz. kapitola Patobiochemie nádorů).
Lyzosomální cesta degradace a její poruchy
Lyzosomy jsou cytoplasmatické organely (průměr 20-500 nm) s kyselým pH (pH 3,8-5,0), které mohou nespecificky degradovat intracelulární (autofagie) i extracelulární proteiny (Obr. 8). Primární lyzosomy vznikají odštěpením z cisteren Golgiho aparátu a části endoplasmatického retikula a obsahují celou řadu hydrolytických enzymů aktivních v kyselém pH. To je v lyzozomech udržováno pomocí protonové pumpy H+-ATPasy. V lyzosomech je přítomno zhruba 50 hydrolytických enzymů, mezi nimiž převažují kathepsiny, ale nachází se zde i řada specifických enzymů, jako jsou kolagenasa, gelatinasa či dipeptidasa.
Hlavní úlohu v degradaci proteinů hrají kathepsiny, které se dělí na několik typů, které patří mezi cysteinové (typ B, C, L, F, H, K, O, S, V, X a W), aspartátové (typ D a E) a serinové (typ A a G) peptidasy. Tyto enzymy se liší buněčnou a tkáňovou distribucí, lokalizací a expresí, biochemickými vlastnostmi (např. pH optimem a specifitou účinku), strukturou a regulací aktivity. Jsou syntetizovány ve formě neaktivních zymogenů, které se aktivují proteolytickým sestřihem v mírně kyselém prostředí endolyzosomu. Kromě nespecifického štěpení proteinů v lyzosomech se některé kathepsiny účastní i specifických buněčných procesů mimo lyzosomy (např. participují na aktivaci kaspas při apoptose, na obnově kostní hmoty, aktivaci proteas a prohormonů, udržení homeostasy epidermis a aktivitě imunitního systému). Aktivita kathepsinů musí být přísně regulována, protože chyby v tomto procesu jsou spojené se vznikem řady patologií (např. nádorová a neurodegenerativní onemocnění, arthritis, osteoporóza). Mezi možnosti řízení jejich aktivity patří regulace exprese, posttranslační modifikace, aktivace zymogenů, pH, působení endogenních a exogenních inhibitorů, nebo kombinace těchto faktorů. pH optimum kathepsinů se nachází v kyselé oblasti (pH ~ 5), takže ani po vylití obsahu lyzosomů do cytoplasmy, kde je neutrální pH, nehrozí natrávení cytosolických proteinů. Hlavními endogenními inhibitory působení kathepsinů mimo lyzosomy jsou proteiny z rodiny cystatinů, serpinů a thyropinů.
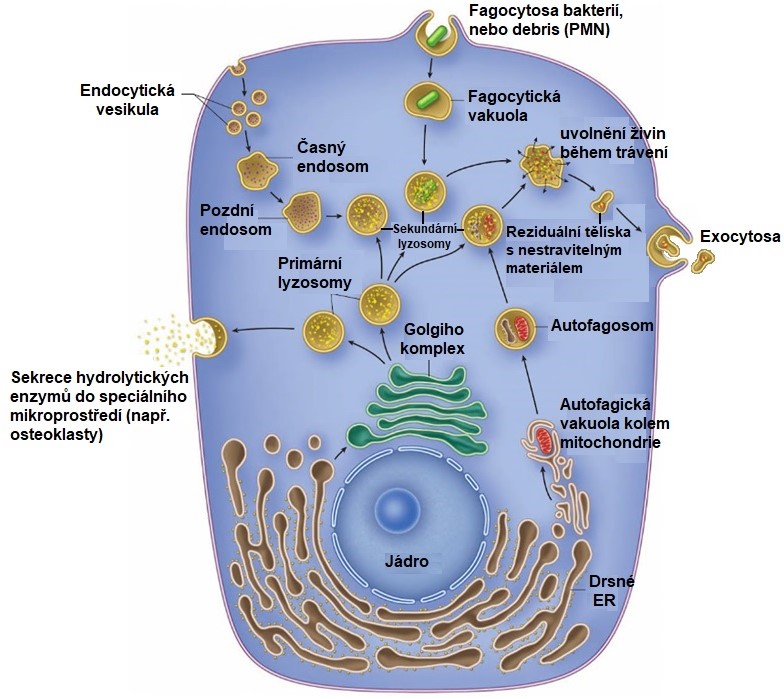
Obr. 8. Vznik lyzosomů a jejich účast v intracelulárním trávicím procesu (upraveno z Mescher 2015)
Poruchy lyzosomální degradace zahrnují jak poruchy se zvýšenou lyzosomální aktivitou, tak poruchy vedoucí ke střádání nerozloženého materiálu vlivem snížené lyzosomální degradace.
Zvýšená lyzosomální degradace může být vyvolána zvýšenou aktivitou lyzosomů (hladovění, diabetes mellitus, úbytek svalové hmoty při denervaci nebo zranění, regrese dělohy po porodu), zvýšeným uvolňováním lyzosomálních enzymů (revmatická arthritis, indukce metastáz u karcinomu mléčné žlázy, kde estrogeny stimulují expresi genu kódujícího kathepsin D), nebo v důsledku mutace fyziologického inhibitoru kathepsinů (Unverrichtova-Lundborgova nemoc, progresivní myoklonická epilepsie, které je způsobená mutací inhibitoru cysteinových kathepsinů - cystatinu B).
V důsledku nedostatečné aktivity některého lyzosomálního enzymu, aktivátoru nebo transportního proteinu se v lyzosomech hromadí nerozložený odpadní materiál, díky tomu se tvoří velké intracelulární vakuoly a rozvíjí se některé z lyzosomálních střádavých onemocnění. Jedná se o skupinu více než 60 progresivních multisystémových onemocnění s autosomálně (vzácně též gonosomálně) recesivní dědičností. Tato onemocnění mají širokou škálu klinických projevů. Závažnost onemocnění závisí na typu hromaděného materiálu a na zbytková aktivitě enzymu/transportéru. Lyzosomální střádavé poruchy můžeme rozdělit do 5 kategorií na defekty v: degradaci glykanů (glykoproteinů, glykolipidů, glykosaminoglykanů a glykogenu), lipidů, proteinů, lyzosomálních transportérech a pohybu lyzosomů („trafficking“). Mezi lyzosomální střádavé poruchy tak řadíme celou řadu mukopolysacharidos a lipidos (např. Gaucherova, Farberova či Niemann-Pickova nemoc), o nichž bylo pojednáno v příslušných kapitolách. Neuronální ceroid-lipofuscinosy jsou skupinou neurodegenerativních lyzosomálních střádavých onemocnění, která je charakterizována progresivní ztrátou mentálních, motorických a zrakových schopností, křečemi a brzkou smrtí. V lyzosomech se hromadí pigmentové inkluze ceroidu a lipofuscinu. Tato onemocnění jsou způsobena mutací genu kódujícího některý z proteinů, který se podílí na lyzosomální degradaci proteinů. Kauzální terapie zatím není známa a prognóza je špatná. Mezi lyzosomální poruchy degradace proteinů patří pyknodysostosis (též Toulouse-Lautrec syndrom) způsobená deficitem kathepsinu K, který se projevuje malým vzrůstem s postižením končetin (z řečtiny dys- = porucha a osteon = kost) a generalizovanou osteosklerosou (z řečtiny pyknos = hustý). Postižení mají krátké a tlusté prsty, potížemi s výměnou zubů a husté, ale křehké kosti. Tímto onemocněním trpěl i slavný francouzský malíř Henri de Toulouse-Lautrec, po němž je tento syndrom rovněž nazýván. Papillonův-Lefèvrův syndrom, vzácný syndrom s palmoplantární hyperkeratosou a těžkou periodontitidou, je způsoben mutací genu pro kathepsin C. Vlivem deficitu kathepsinu C nejsou aktivovány granzymy, serinové peptidasy pocházející z NK-buněk a cytotoxických T‑lymfocytů, které spouštějí apoptosu v cílových buňkách.
Cytosolická cesta degradace a její poruchy
Kvantitativně významnější drahou intracelulárního katabolismu proteinů je tzv. ubikvitin-dependentní proteolýza, která se uskutečňuje v proteasomech. Uplatňuje se např. při regulaci genové exprese proteolýzou specifických chromosomálních proteinů, při regulaci mitotického cyklu, při degradaci abnormálních proteinů a při rozkladu kontraktilních proteinů kosterního svalstva u proteokatabolických stavů. Jedná se o selektivní degradaci probíhající v cytoplasmě za účasti ATP. Nezastupitelnou úlohu v tomto procesu hraje krátký protein ubikvitin (76 aminokyselin) s vysoce konzervovanou primární strukturou (rozdíl mezi ubikvitinem člověka a kvasinky jsou jen 3 aminokyseliny), který je přítomný ve všech eukaryontních buňkách. Ubikvitin se váže na ε-aminoskupiny lysinových zbytků proteinů, které jsou určeny k degradaci. Jeho připojení je katalyzováno specifickými ligasami. K tomu, aby byl protein rozštěpen, musí být k jeho struktuře připojeny minimálně čtyři molekuly ubikvitinu. Takto označený protein je rozpoznán a selektivně degradován v proteasomu, což je makromolekulární komplex složený z podjednotek s různou proteasovou aktivitou.
Při ubikvitinylaci proteinů (Obr. 9) dochází v prvním kroku k aktivaci molekuly ubikvitinu za pomocí ATP (přechodně vzniká ubikvitin-AMP) a jeho připojení na thiolovou skupinu aktivačního enzymu E1. Aktivovaný ubikvitin je následně přenesen na katalytický cystein konjugačního enzymu E2. Komplex E2-ubikvitin je rozeznán enzym E3 ubikvitin-proteinligasou, která vybírá substrát (tedy protein k degradaci) a připojuje na něj ubikvitin. Vznik polyubikvitových řetězců je umožněn opakováním tohoto postupu. Je znám jeden enzym E1, několik desítek enzymů E2 a několik stovek enzymů E3, které mohou ubikvitinylovat tisíce různých substrátů. Za širokou substrátovou specifitu ubikvitinylačního systému je tedy zodpovědný enzym E3. Jednotlivé enzymy E3 mohou spolupracovat jen s některými E2, zatímco proteinové substráty mohou být označeny různými kombinacemi enzymů E2 a E3. Ubikvitinylace je modulována systémem deubikvitinylujících enzymů. Výsledkem je velká specifita a cílené odbourávání jednotlivých proteinů či jejich tříd.
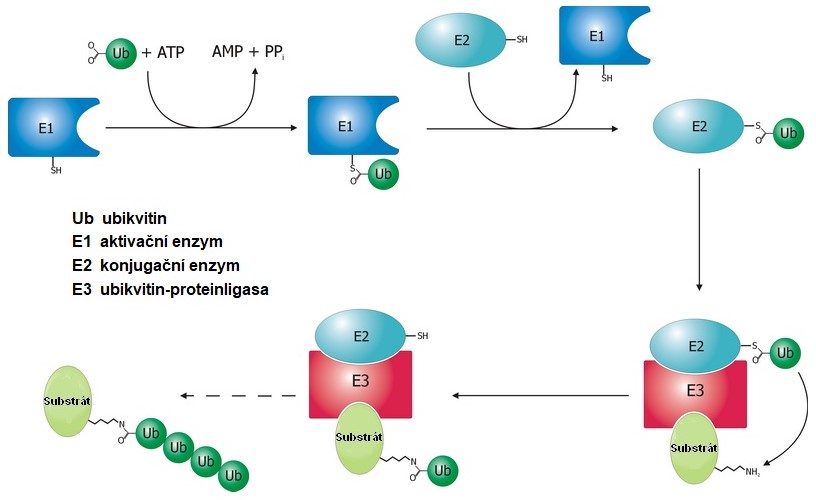
Obr. 9. Mechanismus aktivace a připojení ubikvitinu k proteinu (upraveno z Berg et al. 2006)
Proteolytická degradace označených proteinů probíhá v proteasomu (Obr. 10), což je komplex z velkého množství podjednotek s různou katalytickou aktivitou. Proteasom (26S) je tvořen dvěma subkomplexy – centrální částí (20S, „core particle“) s proteolytickou aktivitou a regulační částí (19S, „regulatory particle“). Uvnitř centrální části, která má soudkovitý tvar a je složená ze čtyř prstenců (dva identické α a dva identické β prstence), dochází k degradaci proteinů. Každý prstenec je tvořený sedmi různými podjednotkami. Na obou koncích centrální části je umístěna regulatorní část, která tvoří jakési víčko/čepičku. Tato část je zodpovědná za rozeznávání polyubikvitinového řetězce, jeho odštěpení, rozvinutí degradovaného proteinu do lineární podoby a jeho vstup do centrální části proteasomu. Molekuly ubikvitinu jsou recyklovány a znovu použity. Rozvolněné a ubikvitinu zbavené substráty jsou pak v centrální části proteasomu postupně hydrolyzovány za spotřeby ATP na peptidy, které jsou po uvolnění do cytosolu hydrolyzovány dalšími enzymy. Za objev tohoto mechanismu degradace proteinů obdrželi v roce 2004 Nobelovu cena za chemii vědci Aaron Ciechanover, Avram Hershko a Irvin Rose.
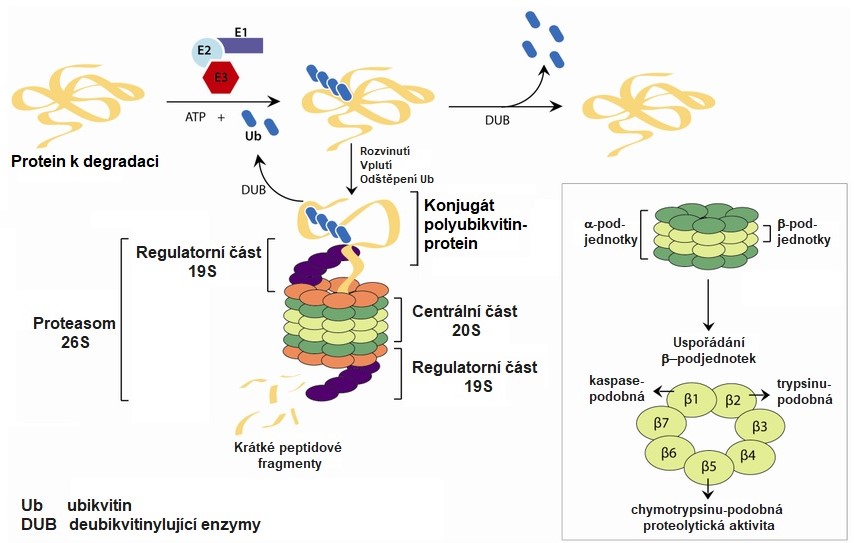
Obr. 10. Ubikvitin-proteasomová cesta degradace proteinů (upraveno z Mattern et al. 2014)
Poruchy ubikvitin-proteasomové cesty (UPS) degradace jsou spojeny mimo jiné se vznikem malignit, neurodegenerativních a renálních onemocnění. Mechanismy, které mohou způsobit změny v ubikvitin-dependentní proteolýze, zahrnují: 1) mutaci nebo deleci substrátu (např. deleci místa pro vazbu ubikvitinu) vedoucí ke zvýšené či snížené degradaci cílového proteinu; 2) mutaci v genech kódujících součásti UPS způsobující změny v jejich expresi; 3) interakci mezi transkripčními faktory, která změní expresi součásti UPS; 4) přítomnost externích faktorů (např. hormony, cytokiny, hypoxie), které regulují aktivitu UPS přímo, nebo prostřednictvím počátečních kroků vedoucích k degradaci substrátu; 5) expresi dalšího faktoru, který urychlí, nebo zpomalí průběh celé dráhy.
Následkem zpomalení ubikvitin-proteasomové degradace proteinů dochází k patologickému hromadění nerozložených proteinů. Příkladem může být Liddleův syndrom, u kterého způsobí bodová mutace v genu kódujícím β nebo γ podjednotku renálního epiteliálního kanálu pro Na+ ionty změnu ve struktuře a aminokyselinovém složení výsledného proteinu. Mutantní protein pak neobsahuje vazebné místo pro ubikvitin-proteinligasu a patologicky se hromadí v membránách renálních buněk. Navíc vede mutace ke zvýšené reabsorpci Na+, která je nezávislá na aldosteronu. Liddleův syndrom se projevuje časným nástupem těžké hypertenze, hypokalemií, metabolickou alkalosou a nízkými plasmatickými hladinami reninu a aldosteronu. Neurodegenerativní onemocnění jsou charakterizována tvorbou deposit agregovaných a chybně složených proteinů v cytoplasmě, jádře, a/nebo v extracelulárním prostoru. Tyto nahromaděné proteiny mohou negativně ovlivnit fungování buňky i tkáně. Příčinou juvenilní Parkinsonovy nemoci je mutace v genu PARK2, který kóduje ubikvitin-proteinligasu (parkin). Mutovaný enzym má pak nižší aktivitu, dochází k hromadění neurotoxických proteinů v inkluzích s následnou degenerací dopaminergních neuronů v substantia nigra. Onemocnění se projevuje časným nástupem příznaků Parkinsonovy nemoci (např. bradykineze, tremor, rigidita, posturální nestabilita). Nádorová onemocnění mohou být vyvolána nedostatečným odbouráváním onkoproteinů (produktů onkogenů), které vyvolávají vznik a podporují růst nádorů. K tomu může dojít, pokud onkoproteiny uniknou degradaci v UPS kvůli mutaci v onkogenu, nebo v některé ze součástí UPS, která je zodpovědná za rozpoznání či ubikvitinylaci onkoproteinu. Patologické nahromadění onkoproteinů podporuje růst a viabilitu nádorových buněk a progresi nádorového onemocnění.
Zrychlené odbourávání proteinů v UPS se projeví nedostatkem funkčního proteinu. Zrychlená degradace tumor-supresorových proteinů může vyvolat vznik nádorového onemocnění. Tumor-supresorový protein p53 hraje významnou úlohu v ochraně buněk před maligní transformací. Onkoprotein E6 z lidského papillomaviru, který je původcem karcinomu děložního čípku, vytváří komplex s lidským p53 a ubikvitin-proteinligasou, a tak podporuje ubikvitinylaci a následnou degradaci proteinu p53. Vysoká exprese některých ubikvitin‑proteinligas je spojená s nádorovými onemocněními, např. u metastáz nemalobuněčného karcinomu plic byla popsána vysoká exprese Skp2, která odbourává inhibitory cyklin-dependentních kinas. Použití inhibitorů proteasomu (např. bortezomibu) je jedním z terapeutických přístupů k léčbě nádorových onemocnění. Bortezomib je prvním lékem cílícím na proteasom, který byl schválen k léčbě mnohočetného myelomu u lidí.