Poruchy intracelulárního metabolismu sacharidů
Příčinou vzniku těchto onemocnění je mutace v genu pro určitý enzym dané metabolické dráhy. Prakticky kterýkoliv krok metabolismu sacharidů může být postižen. K podrobnějšímu popisu jsou vybrány pouze některé významné defekty metabolismu glukosy, fruktosy a galaktosy, které jsou slučitelné se životem (tj. esenciální fruktosurie, dědičná intolerance fruktosy, galaktosemie, deficit glukosa-6-fosfátdehydrogenasy a glykogenosy).
Poruchy v pentosovém cyklu
Pentosový cyklus se skládá ze dvou fází – oxidační a neoxidační. V oxidační fázi dochází k přeměně hexosy (glukosa-6-fosfátu) na pentosu (ribulosa-5-fosfát) za vzniku dvou molekul NADPH. Glukosa-6-fosfát je pomocí glukosa-6-fosfátdehydrogenasy (G6PD) oxidován na 6‑fosfoglukono-δ-lakton za současné redukce NADP+ na NADPH+H+. Tento krok je nevratný a rozhoduje o rychlosti celé dráhy. Vzniklý lakton je laktonasou hydrolyzován na 6‑fosfoglukonát, který je prostřednictvím 6-fosfoglukonátdehydrogenasy dekarboxylován na ribulosa-5-fosfát za současné redukce NADP+ na NADPH+H+. V neoxidační (interakční) fázi je ribulosa-5-fosfát přeměněn zpět na glukosa-6-fosfát sérií isomerizačních a epimerizačních reakcí, kterých se účastní dva enzymy, trasaldolasa a transketolasa. Během těchto reakcí vzniká ribosa-5-fosfát, který je použit k syntéze nukleotidů a nukleových kyselin.
Genetické defekty G6PD s následnou poruchou tvorby NADPH se běžně vyskytují v oblastech kolem Středozemního moře a u lidí afro-karibského původu. Gen pro G6PD je lokalizován na chromosomu X a dosud bylo identifikováno více než 190 různých bodových mutací. Celosvětově je nositelem mutovaného genu pro G6PD asi 400 milionů lidí, což činí z tohoto defektu nejběžnější genetickou vadu. Výsledkem mutace může být vznik nestabilního enzymu (varianta G6PDA-) nebo enzymu, který je stabilní, ale má ve všech erytrocytech nízkou aktivitu (varianta G6PDMediterranean) (Obr. 5).
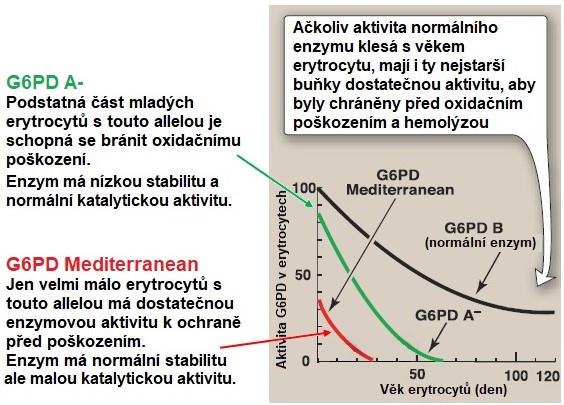
Obr. 5. Pokles aktivity G6PD v erytrocytech u tří běžně se vyskytujících forem tohoto enzymu (upraveno z Harvey a Ferrier 2011).
Erytrocyty jsou bezjaderné buňky, které nejsou schopné proteosyntézy, takže si nemohou doplnit zásobu potřebných proteinů včetně G6PD. Vzhledem k tomu, že jejich funkcí je transport O2 a CO2, jsou často vystaveny tvorbě reaktivních forem kyslíku (ROS). ROS (zejm. H2O2 a organické hydroperoxidy) jsou v těchto buňkách odstraňovány glutathionperoxidasou, která využívá jako kofaktor redukovaný glutathion, i samotným glutathionem. Při odstraňování ROS je glutation oxidován za vzniku oxidovaného glutathionu. K jeho zpětné redukci je potřeba glutathionreduktasa, která používá jako kofaktor právě NADPH. Dalším enzymem, který je závislý na NADPH, je methemoglobinreduktasa, která redukuje Fe3+ v methemoglobinu na Fe2+ za vzniku hemoglobinu. Methemoglobin, který ztrácí schopnost vázat kyslík, vzniká oxidací dvoumocného železa v molekule hemoglobinu na třímocné. V erytrocytech, kde může NADPH vznikat pouze v pentosovém cyklu, způsobí deficit G6PD nedostatek tohoto kofaktoru a díky tomu se sníží i schopnost erytrocytu bránit se proti působení oxidačnímu stresu. Vlivem oxidačního stresu dochází ke změně buněčné struktury erytrocytů, membrány jsou poškozeny lipoperoxidací, hemoglobin precipituje za vzniku Heinzových tělísek (denaturovaný hemoglobin), což se projevuje předčasným rozpadem (hemolýzou) erytrocytů (Obr. 6).
Deficit G6PD se projevuje hemolytickou anemií. Příčinou akutní hemolýzy může být infekce, požití bobů rostliny Vicia faba (vikev bob), užití některých léků (např. antimalarika primachin a pamachin, sulfonamidy, chloramfenikol, ciprofloxacin a další) a kontakt s některými látkami (naftalen). Všechny tyto příčiny zvyšují úroveň oxidačního stresu v erytrocytech. Akutní hemolýza bývá provázena bolestí zad či břicha, sekundárním ikterem v důsledku vzestupu hladiny nekonjugovaného bilirubinu, přechodnou splenomegalií a hemoglobinurií. Hemolýza se objevuje typicky 24 až 72 hodin po požití bobů/léčiva a k úpravě stavu dochází během 4 až 7 dní. Vzácně je hemolýza natolik závažná, že vyžaduje podání krevní transfuze. Oblast výskytu mutovaného genu se přibližně shoduje s oblastí výskytu malárie (a tedy i srpkovité anemie) a heterozygotům poskytuje určitou odolnost vůči malárii.
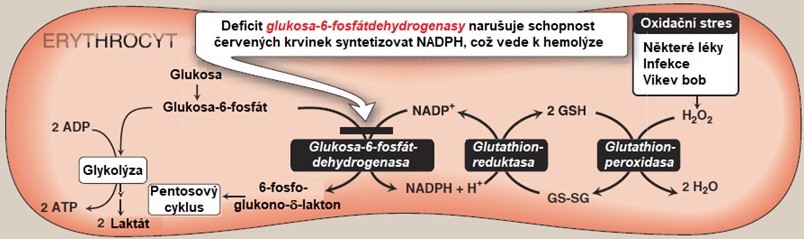
Obr. 6. Následky deficitu G6PD v erytrocytu (upraveno z Harvey a Ferrier 2011).
Poruchy metabolismu fruktosy
V metabolismu fruktosy byly identifikovány dvě dědičné poruchy. Jedná se o esenciální fruktosurii a hereditární intoleranci fruktosy (Obr. 7).
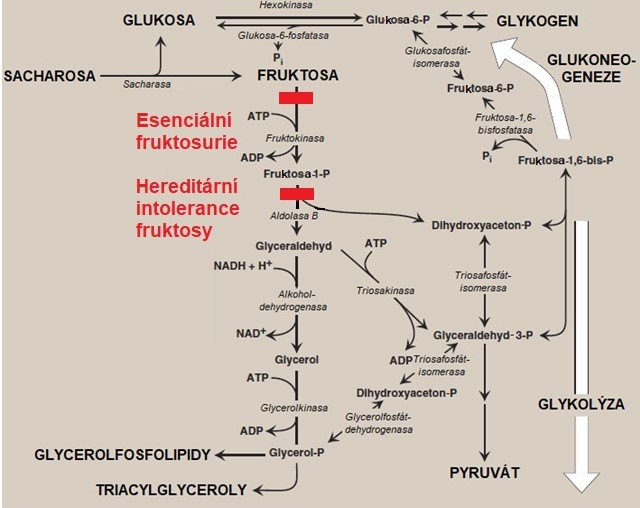
Obr. 7. Metabolismus fruktosy a jeho defekty (upraveno z Harvey a Ferrier 2011).
Esenciální fruktosurie, která je způsobena deficitem fruktokinasy, je neškodná metabolická odchylka. Je charakterizována vylučováním fruktosy močí po požití jídla obsahujícího tento monosacharid. Močí se vyloučí zhruba 10-20 % přijaté fruktosy a zbytek se pomalu metabolizuje alternativními drahami (zejm. hexokinasou ve svalu a v tukové tkáni). Toto onemocnění je děděno autosomálně recesivně. Zavedení dietních opatření není potřeba a prognóza pacientů je výborná.
U hereditární intolerance fruktosy může požití fruktosy vyvolat okamžité gastrointestinální obtíže se zvracením a těžkou hypoglykemii, při trvalém příjmu hrozí selhání jater a ledvin a i.v. podání fruktosy ohrožuje pacienta na životě. Příčinou tohoto onemocnění je deficit aldolasy B, která štěpí fruktosa-1-fosfát na D-glyceraldehyd a dihydroxyacetonfosfát. Díky vysoké aktivitě fruktokinasy se hromadí fruktosa-1-fosfát, který inhibuje glukoneogenezi a glykogenolýzu (alosterická inhibice jaterní fosforylasy), čímž vyvolává hypoglykemii. Navíc vyvolává nadměrnou spotřebu a tím depleci ATP a anorganického fosfátu, který slouží k regeneraci ATP. Díky vyčerpání anorganického fosfátu se ve zvýšené míře tvoří kyselina močová (degradační produkt AMP) a dochází k sérii poruch (např. inhibice proteosyntézy, ultrastrukturní léze), které jsou zodpovědné za poškození funkce ledvin a jater. V důsledku hromadění fruktosa-1-fosfátu dochází k inhibici fosfomanosaisomerasy, která se projeví defekty v glykosylaci proteinů. Terapie sestává z okamžitého vyloučení všech zdrojů fruktosy (fruktosa, sacharóza, sorbitol) ze stravy. Sacharosa by měla být nahrazena glukosou, maltosou a/nebo škrobem. Při dodržování dietních opatření je prognóza výborná s normálním růstem, inteligencí i délkou života.
Poruchy metabolismu galaktosy
Neschopnost metabolizovat galaktosu se vyskytuje u galaktosemií, které mohou být vyvolané dědičnými defekty galaktokinasy, galaktosa-1-fosfáturidyltransferasy nebo UDP‑galaktosa-4-epimerasy (Obr. 8). Všechny defekty jsou děděny autosomálně recesivně.
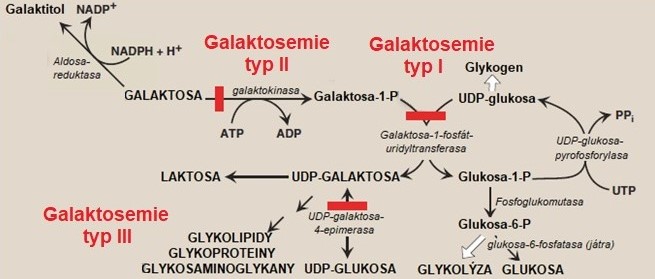
Obr. 8. Metabolismus galaktosy a jeho defekty (upraveno z Harvey a Ferrier 2011).
Defekt galaktosa-1-fosfáturidyltransferasy je neznámější a také nejzávažnější. Označuje se jako typ I či jako klasická galaktosemie. Jedinci s tímto deficitem dokáží fosforylovat galaktosu, ale nejsou schopni metabolizovat galaktosa-1-fosfát. Galaktosa a galaktosa-1-fosfát se hromadí a metabolizují se alternativními drahami na galaktitol a galaktonát. Galaktitol vzniká působením aldosareduktasy v oční čočce a jeho hromadění vyvolává kataraktu již po několika dnech života jedince. Galaktosa-1-fosfát způsobuje v játrech nedostatek anorganického fosfátu, jehož důsledkem je selhání jater a mentální poškození. U kojenců s kompletním či téměř kompletním deficitem se symptomy tohoto onemocnění objevují již několik dní po narození a zahrnují odmítání stravy, zvracení, ikterus a letargii. Později se může přidat hepatomegalie, ascites, otoky a cirrhosa jater. Je zde vysoká četnost neonatálního úmrtí v důsledku sepse vyvolané E. coli. Terapie zahrnuje dietní opatření s okamžitým vyloučením laktosy ze stravy. Pokud je laktosa vyloučena ze stravy velmi časně, symptomy okamžitě mizí a je možné zabránit cirrhose jater.
Deficit galaktokinasy, při kterém není galaktosa fosforylována, je vzácnější formou galaktosemie, ale záludnější než typ I, protože se u něj katarakta rozvíjí bez dalších symptomů intolerance. Galaktosa je z organismu vylučována buď přímo, nebo ve formě galaktitolu (zodpovědný za vznik katarakty). Léčba může být omezena na vyloučení mléka ze stravy, malé zdroje galaktosy lze ignorovat (léky v tabletové formě, mléčné výrobky atd.). Pokud je diagnóza stanovena rychle a léčba zahájena okamžitě, může katarakta úplně vymizet.
Pacienti s deficitem UDP-galaktosa-4-epimerasy nejsou schopni přeměňovat UDP-galaktosu na UDP-glukosu a naopak. Byly popsány dvě formy tohoto onemocnění. Kompletní deficit tohoto enzymu je extrémně vzácný a klinické projevy jsou podobné jako u klasické galaktosemie. Pacienti jsou závislí na příjmu galaktosy potravou, protože si ji nejsou schopni syntetizovat z UDP-glukosy. Při nadměrném příjmu galaktosy potravou se hromadí nezpracovaná UDP-galaktosa a galaktosa-1-fosfát, který vyvolává stejné poškození orgánů jako u klasické galaktosemie. Naopak při nižším příjmu galaktosy, než je aktuální potřeba, se objevují poruchy glykosylace proteinů a lipidů a syntéza glykosaminoglykanů. Léčba je velice obtížná a u dětí s touto poruchou dochází k opoždění psychomotorického vývoje. Častěji se vyskytuje parciální deficit, který je obvykle benigní a nevyžaduje léčebný zásah.
Glykogenosy
Dědičné metabolické poruchy s deficitem aktivity enzymu degradace a syntézy glykogenu nebo transportního proteinu jsou označovány jako glykogenosy. Tato onemocnění jsou charakterizována abnormální strukturou glykogenu, nebo jeho abnormálním obsahem v tkáních. Většina glykogenos je děděna autosomálně recesivně kromě typu IX, kde je dědičnost vázaná na X-chromosom. Glykogenosy jsou (přes určitý překryv) klasifikovány podle zasaženého orgánu na jaterní, svalové a generalizované. Jsou označeny římskými číslicemi, které odrážejí historickou posloupnost jejich objevení, jménem defektního enzymu, nebo jménem autora prvního popisu daného onemocnění (Obr. 9, Tab. 1).
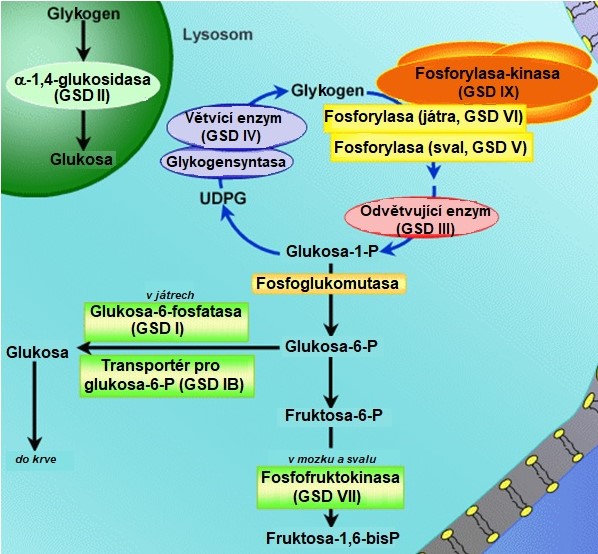
Obr. 9. Glykogenosy (upraveno z Corriel Institute 2018)
Mezi jaterní glykogenosy (GSD) patří von Gierkeho nemoc (GSD I), jaterní forma Coriho nemoci (GSD III), Andersenova nemoc (GSD IV), Hersova nemoc (GSD VI), jaterní formy GSD IX a GSD 0. GSD I, III, VI a IX mají podobné projevy s hypoglykemií, výraznou hepatomegalií a opožděním růstu. Nejzávažnější jaterní glykogenosou je von Gierkeho nemoc (GSD I), která je vyvolána deficitem glukosa-6-fosfatasy. Nahromaděný glukosa-6-fosfát aktivuje glykogensynthasu, takže převládá syntéza glykogenu. Tím, že z glukosa-6-fosfátu nemůže vzniknout glukosa, dochází k těžké hypoglykemii. Zhruba polovina postižených v důsledku choroby zemře. U Coriho nemoci (GSD III, limitní dextrinosa) chybí odvětvující enzym, takže štěpení glykogenu se zastavuje v místech větvení. Okleštěná molekula se nazývá limitní dextrin a má abnormální složení – kratší větve s relativně častějšími vazbami α(1→6). Nemožnost využít části molekuly glykogenu vyvolává mírnou hypoglykemii. Z hlediska přežití má tento typ poměrně příznivou prognózu. Andersenova nemoc (GSD IV, amylopektinosa) je způsobena deficitem větvícího enzymu. Glykogensynthasa není postižena, takže vzniká atypický glykogen s abnormálně dlouhými větvemi a s malým počtem větvících bodů, který připomíná rostlinný amylopektin. Toto onemocnění se manifestuje obvykle v kojeneckém věku nebo v časném dětství jako jaterní selhání s cirrhosou, která vede k terminálnímu postižení jater. Smrt nastává obvykle do dvou let od narození. Hersova nemoc (GSD VI), vyvolaná deficitem jaterní glykogenfosforylasy, a deficit fosforylasa-kinasy (GSD IX) jsou nejméně závažné. Pacienti mají jen mírný sklon k hypoglykemii nalačno a v dospěloti dosahují normální výšky. U GSD 0 je kvůli deficitu glykogensynthasy postižena syntéza glykogenu. Onemocnění se projevuje v kojeneckém věku a v časném dětství hypoglykemií a ketosou nalačno a postprandiální hyperglykemií a hyperlaktacidemií.
Léčba jaterních glykogenos je primárně dietní a jejím cílem je zabránit vzniku hypoglykemie a potlačit sekundární metabolickou dekompenzaci. To obvykle vyžaduje časté krmení přes den a u typů GSD I a GSD III i kontinuální noční krmení nasogastrickou sondou.
Svalové glykogenosy jsou charakterizovány intolerancí cvičení s myalgií a svalovými křečemi vyvolanými cvičením, které jsou často provázeny následnou rhabdomyolýzou a myoglobinurií. Patří mezi ně McArdlova nemoc (GSD V), Taruiho nemoc (GSD VII), XSD X, GSD XI, GSD XII, GSD XIII a GSD XV. Všechny symptomy v klidu mizí. McArdlova nemoc (GSD V) je způsobena deficitem svalové glykogenfosforylasy. Svaly tedy nedokáží svůj glykogen využít a nejsou schopny většího výkonu. Tato choroba je relativně mírná. U Taruiho nemoci chybí svalová fosfofruktokinasa. Tento deficit brání volnému průběhu glykolýzy ve svalech, hromadí se fruktosa-6-fosfát a glukosa-6-fosfát, což vede ke zvýšené syntéze glykogenu ve svalech. Ostatní zmíněné typy GSD jsou velmi vzácné a nebudeme je dále rozebírat. V případě svalových glykogenos neexistuje specifická léčba.
Mezi generalizované glykogenosy patří Pompeho nemoc (GSD II), která je provázena deficitem lyzosomální α-glukosidasy (též kyselé maltasy). Na rozdíl od ostatních glykogenos, u kterých dochází ke střádání glykogenu v cytoplasmě, patří Pompeho nemoc mezi lyzosomální střádavé choroby. Normálně je glykogen, který se dostane do lyzosomu, degradován na glukosu, která může difundovat přes membránu, zatímco u tohoto onemocnění se lyzosomy plní glykogenem. Mimo lyzosomy je metabolismus glykogenu normální. Choroba má několik různě závažných forem, z nichž nejtěžší juvenilní forma končí smrtí již v prvním roce života. Onemocnění je vždy provázeno svalovou slabostí a často končí kardiopulmonálním či respiračním selháním. K léčbě GSD II může být využita enzymová substituční terapie s použitím rekombinantní lidské α-glukosidasy. Paliativní léčba zahrnuje podporu dýchání, aerobní cvičení a stravu bohatou na proteiny.
Tab. 1. Přehled glykogenos a jejich hlavní klinické příznaky
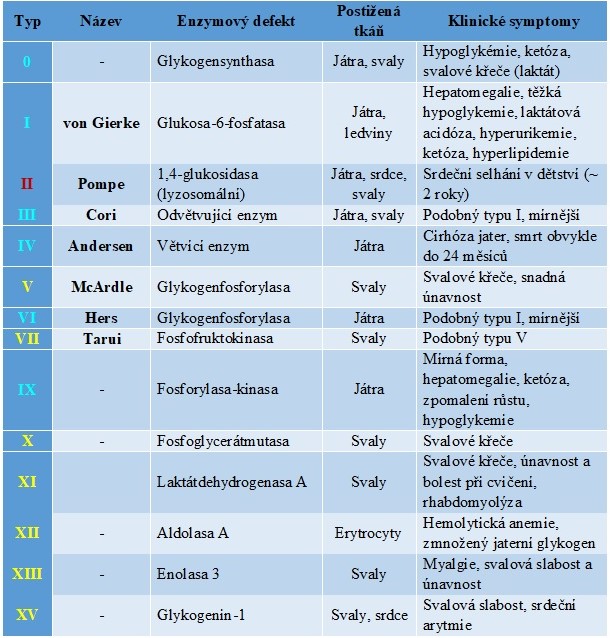
Svalové glykogenosy (žlutě označené), jaterní glykogenosy (modře označené), generalizované glykogenosy (červeně označené)