Diabetes mellitus
Jedná se o chronické heterogenní onemocnění provázené hyperglykemií v důsledku dlouhodobého absolutního nebo relativního nedostatku insulinu nebo jeho nedostatečného účinku ve tkáních. Následkem nedostatku insulinu se rozvíjí komplexní metabolická porucha, která zasahuje do metabolismu sacharidů, lipidů a proteinů i do hospodaření s vodou a elektrolyty. V současné době se DM podle American Diabetes Association klasifikuje na čtyři typy: 1) DM 1. typu; 2) DM 2. typu; 3) další specifické typy DM (genetické defekty β-buňky či účinku insulinu, onemocnění exokrinního pankreatu, endokrinopatie – hormony regulace glykemie, lékové a chemické vlivy, infekce, neobvyklé formy imunitně podmíněného DM, jiné genetické syndromy někdy provázející DM); a 4) gestační DM.
Diabetes mellitus 1. typu
DM1 (též označován jako DM1A) je vyvolán autoimunitní destrukcí β-buněk pankreatu, která způsobí absolutní deficit insulinu. Tento typ DM se poprvé manifestuje již v dětství a je nutná celoživotní léčba insulinem. U jednovaječných dvojčat je 30-50% šance, že se tento typ DM objeví u obou sourozenců. Charakteristickými znaky DM1 je rychlý nástup těžké hyperglykemie, úbytek hmotnosti a diabetická ketoacidosa, která může být smrtelná.
K vývoji DM1 je třeba genetická predispozice, která umožní rozpoznání β-buněk jako něčeho cizorodého, a vnější stimul (expozice viru či toxinu). Právě expozice viru/toxinu může u geneticky predisponovaných jedinců spustit destrukci β-buněk T-lymfocyty a makrofágy, která je provázena insulitidou (zánětem) nejprve bez poruchy sekrece insulinu. V krvi pacientů s DM1 jsou přítomné protilátky proti β-buňkám a jejich součástem (protilátky IAA proti insulinu, ICA proti buňkám Langerhansových ostrůvků, GAD proti glutamátdekarboxylase v Langerhansových ostrůvcích), které mají diagnostický význam. V průběhu let jsou postupně ničeny β-buňky a dochází tedy ke snížení tvorby insulinu. Když kapacita pankreatu pro tvorbu insulinu klesne pod prahovou hodnotu (je zničeno 80-90 % β-buněk), objeví se náhle příznaky DM1 a pacient je odkázán na terapii insulinem.
V některých afrických a asijských populacích je popisován idiopatický diabetes s neznámou etiologií. U tohoto typu označovaného také jako DM1B jsou β-buňky zničeny, i když nejsou v krvi přítomné autoprotilátky.
Metabolické změny při DM1
DM1 je typické katabolické onemocnění, které se manifestuje hyperglykemií, glykosurií, ketoacidosou, ketonurií, dyslipidemií, zvýšenou hladinou laktátu, deplecí tukové tkáně a katabolismem proteinů. Patogeneze těchto změn souvisí s nedostatkem insulinu a zvýšenou tvorbou antagonistů insulinu (glukagon, katecholaminy a kortisol). Důsledkem je převaha katabolických reakcí nad reakcemi anabolickými (Obr. 16).
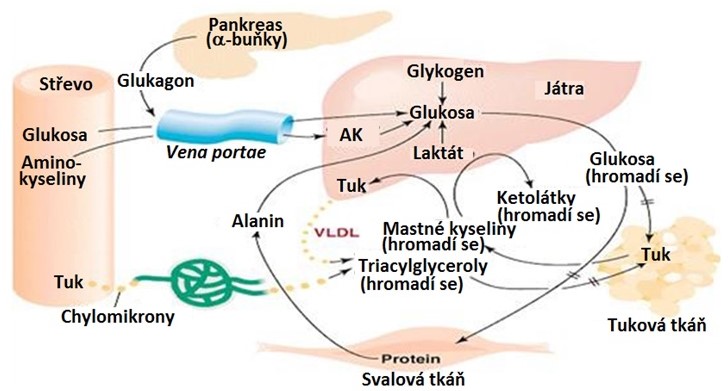
Obr. 16. Hlavní změny v metabolismu živin u neléčeného DM 1. typu (upraveno z Devlin 1997)
Orgány zůstávají i při dostatečném přísunu živin ze střeva v katabolické fázi. Charakteristickou změnou je hyperglykemie, která se může až desetinásobně zvýšit v porovnání s normálními hladinami (~ 5 mmol/l). Příčinou vzniku hyperglykemie je jednak snížení příjmu glukosy do insulin-dependentních tkání (tuková tkáň, srdeční a kosterní svalstvo), protože vlivem nedostatku insulinu nejsou tyto tkáně schopné vystavit na svých membránách transportér GLUT4. Dalším důvodem je inhibice glykolýzy (inhibice klíčových enzymů a pyruvátdehydrogenasy) a aktivace glukoneogeneze (aktivace klíčových enzymů), glykogenolýzy (inhibice glykogensynthasy a aktivace glykogenfosforylasy) a uvolnění glukosy z jater do krve. Díky inhibici glykolýzy a syntézy glykogenu nemůže být glukosa vytvořená v glukoneogenezi v játrech využita a je i přes vysokou koncentraci v extracelulární tekutině transportována do krve. Pokud hladina glukosy v krvi překročí transportní kapacitu ledvin, pak se glukosa objevuje v moči (glykosurie), což se projeví osmotickou diurézou a rychle se rozvíjející dehydratací, pokud není adekvátně zvýšen příjem tekutin.
Díky nízkému poměru insulin/glukagon je v tukové tkáni inhibována lipoproteinová lipasa a aktivována hormon-sensitivní lipasa, což vede k nekontrolované lipolýze a do krve se dostává velké množství mastných kyselin. Jejich oxidace v řadě tkání inhibuje oxidaci glukosy (tzv. Randleův cyklus), protože vznikající meziprodukty (citrát, acetyl-CoA, NADH) přímo inhibují metabolické dráhy oxidace glukosy (pyruvátdehydrogenasa, fosfofruktokinasa), což vyvolá nahromadění glukosa-6-fosfátu, který inhibuje hexokinasu a následně dojde k omezení schopnosti svalových buněk přijímat a zpracovávat glukosu. Játra zpracovávají mastné kyseliny částečně na triacylglyceroly, které jsou ve formě VLDL secernovány do krve. Jejich zpracování však kvůli inhibici lipoproteinové lipasy vázne a rozvíjí se hypertriacylglycerolemie. Při β‑oxidaci mastných kyselin vzniká velké množství acetyl-CoA, který nemůže všechen vstoupit do citrátového cyklu (nedostatek oxalacetátu kvůli probíhající glukoneogenezi), je jeho část převedena do syntézy ketolátek a cholesterolu. Ketolátky se vylučují především močí, část acetonu se eliminuje plícemi. Při vysokých koncentracích ketolátek dojde k překročení kapacity ledvin a ketolátky se objevují v moči (ketonurie). Zároveň dojde k poruše acidobazické rovnováhy, protože ketolátky mají povahu středně silných kyselin a působí tedy při vysokých koncentracích jako pufry, takže mohou vyčerpat alkalickou zásobu a pak dojde k rozvoji ketoacidosy. Nahromadění kyselin a vznikající acidosa nutí pacienty k hlubokému dýchání (Kussmaulovo dýchání).
Celkový obrat proteinů u neléčeného DM1 v důsledku zvýšeného proteokatabolismu a snížené syntézy proteinů stoupá a zvyšují se ztráty dusíku močí. Rozkladem proteinů je postiženo hlavně kosterní svalstvo. Jedním z hlavních energetických substrátů pro sval se stávají aminokyseliny s větveným řetězcem (stoupá aktivita komplexu dehydrogenas 2‑oxokyselin s větveným řetězcem). S aktivací proteolýzy ve svalech a degradací aminokyselin s větveným řetězcem je spojena zvýšená syntéza alaninu a glutaminu. Většina aminokyselin uvolněných svalovými buňkami do krve je využita ve viscerálních tkáních jako energetický zdroj a jako substrát pro syntézu glukosy.
Diabetes mellitus 2. typu
Jedná se o nejčastější formu DM (až 90 % z celkového počtu diabetiků) a manifestuje se obvykle u starších a obézních osob. V posledních letech se však významně zvýšil výskyt DM2 u obézních adolescentů. V tomto případě je nedostatek insulinu relativní. DM2 se rozvíjí pomalu, často po řadu let. Genetické vlivy jsou zde silnější než u DM1, jednovaječná dvojčata mají až 72% pravděpodobnost, že budou obě postižená touto nemocí. DM2 je charakterizován hyperglykemií, glykosurií a dyslipidemií podobného charteru jako DM1. Hladina ketolátek je však obvykle normální a nedochází ani k rozvoji proteokatabolismu. Většina metabolických odchylek pozorovaných u tohoto typu DM je vyvolána rezistencí tkání k insulinu.
Insulinová rezistence je stav, při němž fyziologické množství insulinu vyvolá subnormální biologickou odpověď. Je způsobena sníženým počtem receptorů nebo jejich necitlivostí k insulinu. Na vzniku insulinové rezistence se podílejí vlivy vrozené (např. mutace insulinového receptoru, transportérů pro glukosu či signální proteiny) a vlivy získané (např. obezita, vysoká koncentrace volných mastných kyselin, přejídání, stáří, medikace). Genetické defekty mohou být pre-receptorové (např. abnormální insulin, protilátky proti insulinu), receptorové (např. menší počet receptorů, omezená schopnost vázat insulin, mutace receptoru, jeho zablokování protilátkami) či post-receptorové (např. poruchy v přenosu signálu, mutace GLUT4). Nejčastější příčinou však je obezita, do jejíhož průběhu se významně zapojují hormony produkované tukovou tkání, tzv. adipokiny (leptin, adiponektin, rezistin, visfatin a další). Pokud jsou β-buňky schopné produkovat insulin, pak dochází při zvýšených požadavcích organismu na insulin ke zvýšení jeho produkce (hyperinsulinemie).
Metabolické změny při DM2
U DM2 je insulin přítomen, ale tkáně jsou vůči němu méně vnímavé. Charakteristickým znakem DM2 je hyperglykemie, která vzniká v důsledku sníženého příjmu glukosy do periferních tkání (hlavně buněk kosterního svalstva) a zvýšené jaterní glukoneogeneze. Dalším průvodním znakem tohoto onemocnění je hypertriacylglycerolemie, která vzniká v důsledku rychlé de novo syntézy mastných kyselin a VLDL v játrech spíše než kvůli zvýšené dodávce mastných kyselin z tukové tkáně (DM1). Nekontrolované lipolýza totiž není u tohoto typu DM přítomná a díky přítomnosti insulinu nevzniká ani ketoacidosa (Obr. 17).
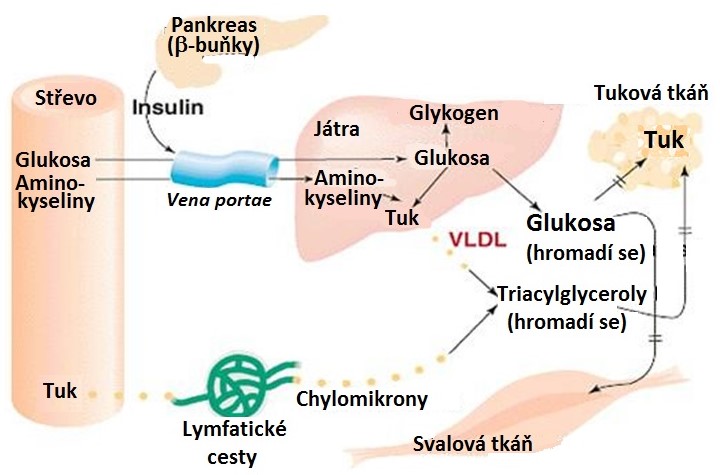
Obr. 17. Hlavní změny v metabolismu živin u neléčeného DM 2. typu (upraveno z Devlin 1997)
Na patogenezi insulinové rezistence se podílejí i volné mastné kyseliny (VMK), protože obézní jedinci a pacienti s DM2 mají vyšší hladiny VMK než zdraví jedinci. Při zvýšené nabídce VMK dochází v tkáních k jejich přednostní oxidaci a k inhibici drah oxidujících glukosu (Randleův cyklus). Při nízké aktivitě svalové hexokinasy se omezuje schopnost svalové buňky přijímat a zpracovávat glukosu a rozvíjí se insulinové rezistence na úrovni svalové buňky. Kromě toho dochází ve svalové buňce k nahromadění meziproduktů oxidace mastných kyselin (acyl-CoA s dlouhým řetězcem), které inhibují enzymy syntézy glykogenu a utilizace glukosy. Tyto meziprodukty mohou přeměněny též na diacylglycerol (DAG), významnou signální molekulu, který aktivací proteinkinasy C vyvolává abnormální fosforylaci IRS a tím inhibici signalizační dráhy insulinového receptoru a tím i snížení transportu glukosy do svalové buňky. V játrech je v důsledku zvýšené nabídky VMK aktivována glukoneogeneze bez současné inhibice glykogenolýzy (autoregulační mechanismus u zdravých osob) a játra tak uvolňují glukosu do krve v situaci, kdy nejsou svalové buňky schopné tento nadbytek glukosy přijmout. Pokud má být za této situace udržena normální hladina glukosy, musí se zvýšit tvorba insulinu v pankreatu. Při krátkodobém (do 6 hodin) vystavení β-buněk pankreatu působení zvýšených hladin VMK dochází ke zvýšení glukosou-stimulovaného výdeje insulinu. Při dlouhodobé expozici VMK však dochází ke snížení produkce insulinu (asi díky navození oxidačního stresu v β-buňkách), tak ke snížení periferní vnímavosti tkání k insulinu. V této situaci je porušena regulace glykemie, rozvíjí se porucha glukosové tolerance a DM2.
Komplikace diabetu mellitu
U obou typů diabetu dochází k řadě komplikací, které vznikají v návaznosti na popsané metabolické změny. Tyto komplikace můžeme rozdělit na akutní a chronické. Mezi akutní komplikace DM patří hypoglykemické kóma, hyperglykemické diabetické kóma s ketoacidosou, hyperosmolární kóma bez ketoacidosy a laktacidotické kóma. Chronické komplikace DM, které můžeme rozdělit na mikro- a makrovaskulární komplikace, jsou představovány degenerativními změnami krevních a nervového systému.
Akutní komplikace diabetes mellitus
Při poklesu glykemie pod 3,3 mmol/l hovoříme o hypoglykemii. Hypoglykemie se vyskytuje častěji u pacientů s DM1. Zhruba jedna třetina diabetiků léčených insulinem prodělá alespoň jednou v životě hypoglykemické kóma a 2-4 % pacientů s DM1 na hypoglykemii zemřou. Příčinou vzniku hypoglykemie je nejčastěji podání příliš velké dávky insulinu, nedostatečný příjem potravy nebo nadměrná či nepředpokládaná fyzická zátěž. Při hypoglykemii dochází nejprve k aktivaci kontraregulačních mechanismů, která se projeví neklidem, třesem, pocením, zčervenáním, tachykardií a pocitem hladu. Při pokračující hypoglykemii se rozvíjí symptomy porušené funkce CNS (neuroglykopenie) – snížení psychomotorických a intelektuálních funkcí, které jsou při dalším poklesu glykemie provázeny poruchami vědomí až kómatem. Akutní léčba sestává z p.o. podání 10-20 g volných sacharidů (tj. 2-4 kostky cukru), které lze po 5-10 minutách opakovat. U pacientů s poruchou vědomí se podává 20% glukosa i.v. a glukagon i.m.
Diabetická ketoacidosa je nejčastější příčinou úmrtí diabetiků do 20 let věku, mortalita je kolem 5 %. Častěji se vyskytuje u pacientů s DM1. Je provázena hyperglykemií (≥ 15 mmol/l), která je příčinou vystupňované osmotické diurézy vyvolávající dehydrataci pacienta. Dehydratace vede k hyperosmolaritě, která může vést až k poruše vědomí a kómatu. Metabolická acidosa se rozvíjí díky vystupňované syntéze ketolátek v játrech (ketonemie > 5 mmol/l) a je často provázena hyperventilací (Kussmaulovo dýchání). Příčinou vzniku tohoto stavu je absolutní nebo relativní nedostatek insulinu či nadbytek kontraregulačních hormonů. Při léčbě se doplňují chybějící tekutiny, insulin, minerály, upravuje se vnitřní prostředí a intenzivně se léčí vyvolávající příčina (např. podání antibiotik u infekcí, koronární intervence u infarktu myokardu).
Hyperosmolární syndrom bez ketoacidosy je akutní komplikací především DM2 s vysokou mortalitou (15-30 %). Projevuje se extrémní hyperglykemií (> 35–50 mmol/l) s těžkou dehydratací a hyperosmolaritou plasmy, rizikem vzniku prerenální renální insuficience různého stupně a poruchami vědomí. Ketoacidóza není přítomná. Mezi nejčastější příčiny vzniku patří kardiovaskulární příhody, rozsáhlejší infekce či nepřiměřená terapie některými léčivy (diuretika, kortikoidy, β-blokátory), které znemožní nemocnému dostatečný příjem tekutin při osmotické diuréze ze stoupající glykemie. V patogenezi se uplatňuje relativní nedostatek insulinu s nadbytkem kontraregulačních hormonů. Léčebná opatření zahrnují vždy hospitalizaci na jednotce intenzivní péče, i.v. podání tekutin, insulinu, korekce mineralogramu, komplexní léčba šokového stavu a vyvolávající příčiny (antibiotika, antikoagulační léčba atd.).
Laktátová acidosa je metabolická acidosa (pH < 7,35) se zvýšenou hladinou laktátu v krvi (> 6 mmol/l), která pacienta ohrožuje na životě a má značnou mortalitu (až 50 %). Jedná se o vzácnou komplikaci pacientů s DM2 léčených biguanidy (zejm. fenformin a buformin). Objevuje se u nemocných, u nichž nebyly dodrženy kontraindikace léčby biguanidy. Projevy jsou často nespecifické, a pokud není laktátová acidosa včas rozpoznána, rozvíjí se těžká metabolická acidosa se vzestupem laktátu a se závažnými změnami celkového stavu pacienta, které často končí smrtí dotyčného. Léčba probíhá na jednotce intenzivní péče za použití hemopurifikačních metod.
Chronické komplikace diabetes mellitus
Při dlouhodobém působení hyperglykemie dochází po letech až desetiletích k nevratným změnám v organismu, které označujeme jako chronické komplikace DM. Ty můžeme rozdělit na makrovaskulární a mikrovaskulární a hovoříme o diabetické mikro- a makroangiopatii. Diabetická makroangiopatie (ischemická choroba srdeční, cévní mozkové příhody, ischemická choroba dolních končetin) je souhrnné označení pro aterosklerotické projevy na velkých tepnách diabetiků. Diabetická atherosklerosa je charakteristická časnějším vznikem a rychlejší progresí. Mezi klasické mikrovaskulární komplikace patří diabetická nefropatie, retinopatie a neuropatie.
V rozvoji mikro- i makrovaskulárních komplikací hraje významnou úlohu oxidační stres. Z dlouhodobých výzkumů chronických komplikací diabetu vyplývá, že hlavní příčinou metabolických abnormalit pozorovaných u DM je nadprodukce reaktivních forem kyslíku (ROS) mitochondriemi. Nadbytek ROS amplifikuje další tvorbu ROS aktivací NADPH‑oxidas a endoteliální synthasy NO. Stabilní ROS difundují do jádra, kde poškozují DNA, čímž je aktivována poly(ADP-ribosa)polymerasa (PARP). Ribosylace glyceraldehyd-3-fosfátdehydrogenasy (GAPDH) katalyzovaná PARP snižuje aktivitu GAPDH, což vyvolá hromadění časných meziproduktů glykolýzy, které se následně přesouvají do čtyř patogenních signálních drah. Díky inhibici GAPDH stoupá koncentrace glyceraldehyd-3-fosfátu (GAP), který je metabolizován na methylglyoxal, jeden z velmi reaktivních meziproduktů neenzymové glykace (viz. dále). Isomerizací GAP vzniká dihydroxyacetonfosfát, který může být metabolizován na DAG, významný aktivátor proteinkinasy C. Fruktosa-6-fosfát může být přesměrován do hexosaminové dráhy, kde z něj vzniká UDP-N-acetylglukosamin (UDP‑GlcNAc), který působí jako ligand některých transkripčních faktorů a mění transkripci specifických genů. Nadbytek glukosy jev polyolové cestě přeměněn aldosareduktasou na cukerný alkohol sorbitol, který díky zvýšení osmotického tlaku v buňce vyvolává poškození oční čočky, nervů a glomerulů ledvin. Cesty tkáňového poškození vyvolané hyperglykemií jsou shrnuty v Obr. 18.
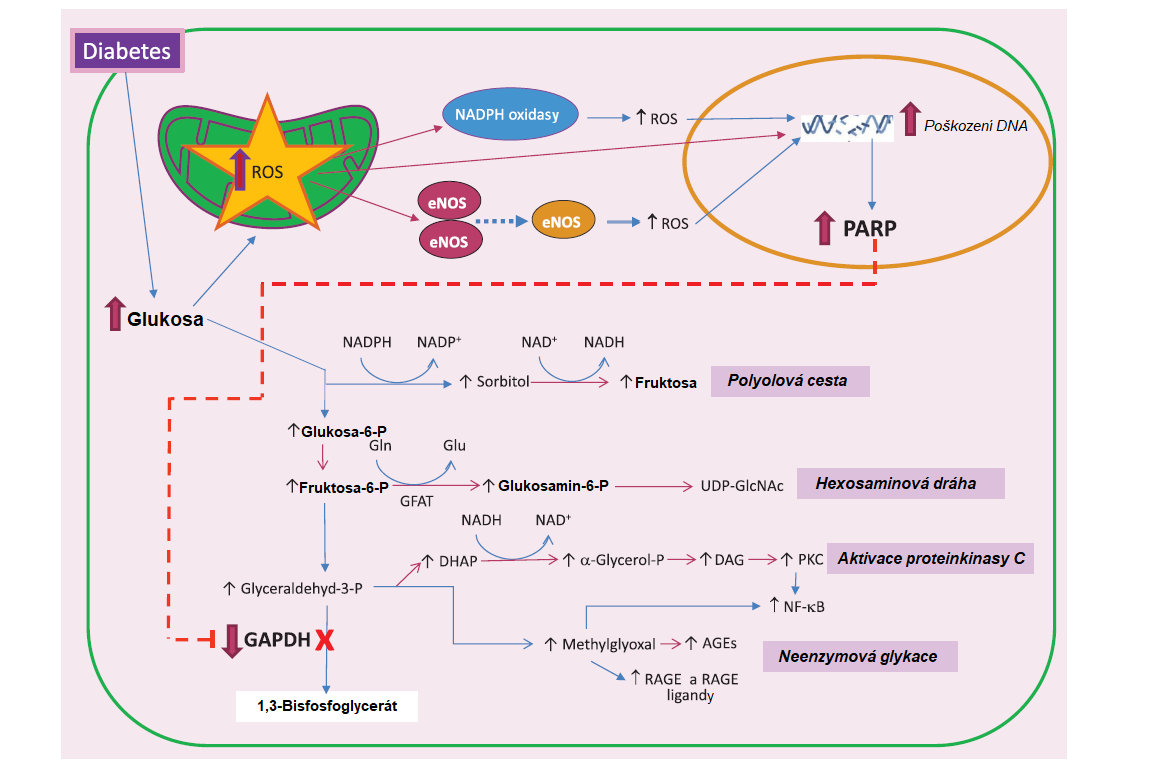
Obr. 18. Čtyři patogenní mechanismy indukované hyperglykemií jsou aktivovány nadprodukcí ROS (upraveno z Shah a Brownlee 2016). AGEs, koncové produkty pokročilé glykace; DAG, diacylglycerol; DHAP, dihydroxyacetonfosfát; eNOS, endoteliální synthasa oxidu dusnatého; GAPDH, glyceraldehyd-3-fosfátdehydrogenasa; GFAT, glutamin:fruktosa-6-fosfát-amidotransferasa; Glu, glutamát; Gln, glutamin; NF-κB, nukleární faktoru κB; PARP, poly(ADP-ribosa)polymeráza; PKC, proteinkinasa C; RAGE, receptor pro AGE; ROS, reaktivní formy kyslíku; UDP-GlcNAc, UDP-N-acetylglukosamin
Polyolová cesta
Glukosa se v buňkách, které obsahují aldosareduktasu (např. oční čočka, neurony, glomeruly), redukuje na cukerný alkohol sorbitol (Obr. 19). Tato dráha je aktivována hyperglykemií, protože KM aldosareduktasy pro glukosu je vysoké (70 mmol/l). Sorbitol nemůže díky své silně hydrofilní povaze přecházet přes buněčné membrány, v buňce se hromadí a osmoticky přitahuje vodu, což vyvolává osmotický edém buňky. V případě oční čočky vyvolává edém denaturaci proteinů krystalínů, které vytvářejí ložiska rozptylující světlo, a tak vzniká zákal čočky (katarakta). Reakce katalyzovaná aldosareduktasou spotřebovává NADPH, který je nezbytný např. k udržení hladiny redukovaného glutathionu (významný antioxidant) v buňce, takže jsou buňky méně chráněné před oxidačním stresem. Při zvýšení poměru NADP+/NADPH se rozvíjí diabetická pseudohypoxie. Aktivita polyolové cesty může rovněž přispívat k depleci NAD+, protože sorbitol je sorbitoldehydrogenasou (SDH) oxidován za účasti NAD+ na fruktosu. Díky spotřebě NAD+ kompetuje SDH s GAPDH, díky čemuž se v buňce hromadí triosafosfáty, které se mohou stát prekurzory tvorby AGEs. Vznikající fruktosa pak může rovněž přispět k tvorbě AGEs, protože je nejprve fosforylována na fruktosa-3-fosfát, který je degradován na 3-deoxyglukoson. Oba tyto produkty jsou silnými glykačními činidly a podporují vznik AGE produktů.
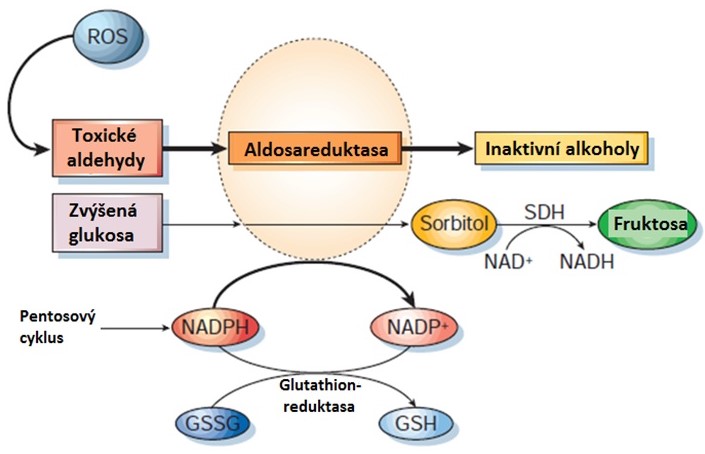
Obr. 19. Aldosareduktasa a polyolová cesta (upraveno z Brownlee 2001). GSH, redukovaný glutathion, GSSG, oxidovaný glutathion
Neenzymová glykace (tvorba AGE produktů)
Neenzymová glykace (též Maillardova reakce) patří mezi posttranslační modifikace proteinů. Probíhá na volných aminoskupinách proteinů, fosfolipidů a nukleových kyselin, kde dochází k navázání molekul redukujících sacharidů. Ty jsou sledem následných reakcí přeměněny až na koncové AGE produkty. Glykace probíhá ve všech organismech kontinuálně během celého života a významnou měrou se podílí na změnách provázejících stárnutí organismu. Všichni jsme tedy v průběhu života vystaveni působení glukosy, a čím vyšší je její koncentrace v krvi, tím závažnější jsou následky. Neenzymová glykace se významně podílí na patogenezi a progresi celé řady onemocnění včetně DM.
Neenzymová glykace je komplexní proces (Obr. 20), který je v časných fázích závislý na koncentraci glukosy. Probíhá pomalu, týdny až měsíce, a je zahájen kondenzací redukujícího cukru nebo podobné sloučeniny (např. askorbátu) s volnou aminoskupinou proteinu, lipidu nebo nukleové kyseliny. V případě glukosy vzniká v rozmezí několika hodin nestabilní aldimin známý jako Schiffova báze, který přesmykem poskytuje relativně stabilní ketoamin, tzv. Amadoriho produkt (např. fruktosamin). Tvorba Amadoriho produktu je do určité míry vratným procesem, avšak rovnováha reakce je výrazně posunuta ve prospěch jeho vzniku. Klasickým přesmykem je Amadoriho produkt postupně přeměněn na AGEs a to buď za neúčasti, nebo při účasti kyslíku (glykoxidace). Schiffova báze i Amadoriho produkt jsou ve střední fázi glykační reakce degradovány oxidačními a dehydratačními reakcemi na řadu α‑dikarbonylových sloučenin (např. glyoxal, methylglyoxal a 3-deoxyglukoson). Rovněž autooxidace glukosy katalyzovaná ionty přechodných kovů je zdrojem dikarbonylových sloučenin. Ty reagují s proteiny ochotněji než parentní monosacharid a vytvářejí různé zkřížené vazby a AGE produkty. Kromě dikarbonylových sloučenin vznikají rovněž ROS (např. peroxid vodíku), které iniciují další oxidační kroky a podílejí se tak na vzniku AGE produktů. Během týdnů až měsíců vznikají v pozdní fázi koncové produkty pokročilé glykace. Jejich vznik je urychlen přítomností oxidačního a karbonylového stresu.
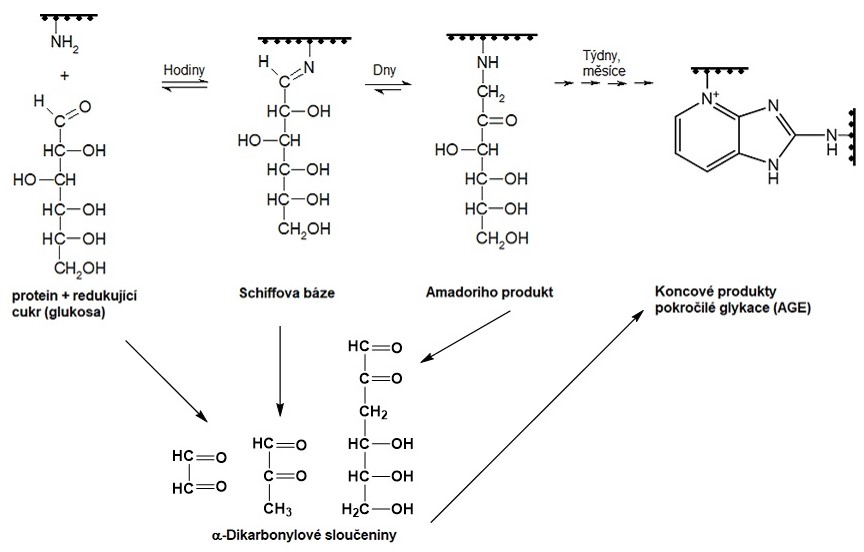
Obr. 20. Průběh glykační reakce
AGE produkty tvoří heterogenní skupinu látek, pro něž je charakteristická žlutohnědá pigmentace, fluorescence, schopnost vzájemného zesítění (crosslinking) a reakce s receptorem RAGE, který je specifický pro AGEs. Existují ovšem i AGE nefluoreskující a AGE, které schopnost zesítění nemají. Struktura řady AGE není známa, ale některé z nich byly popsány jako modelové AGE a další jsou postupně charakterizovány. Zřejmě nejvýznamnějším AGE produktem schopným tvořit zesítění je pro člověka glukosapan, jehož tkáňová koncentrace až tisícinásobně převyšuje koncentrace dalších AGE produktů schopných tvořit zesítění. Kromě endogenní tvorby AGEs pocházejí také z exogenních zdrojů (např. potrava, tabákový kouř).
Prekursory AGE produktů mohou být v organismu detoxifikovány pomocí specifických reduktas (např. glyoxalasou). AGE-modifikované proteiny jsou vychytány makrofágy díky scavengerovým receptorům na jejich povrchu. Tyto proteiny jsou intracelulárně degradovány a uvolněné malé rozpustné AGE-peptidy a AGE-adukty jsou vyloučeny ledvinami. Snížená funkce ledvin či poškození funkce jater může vést ke zvýšení jejich plazmatických koncentrací a k hromadění v tkáních, které má řadu toxických účinků. AGE mohou přímo poškodit strukturu mezibuněčné hmoty, změnit její fyzikální a chemické vlastnosti, metabolismus a způsobit abnormální zesítění, blokovat vasodilatační působení oxidu dusnatého, indukovat lipoperoxidaci nebo působit přes specifické receptory. Těch bylo popsáno několik a nejvýznamnějším z nich je RAGE. Jeho aktivace vede ke spuštění signální kaskády zahrnující aktivaci NF-κB, která je spojena s oxidačním stresem. Následuje stimulace transkripce genů pro prozánětlivé cytokiny, růstové faktory a adhezivní molekuly.
Glykace proteinů s sebou přináší celou řadu následků pro jejich strukturu a aktivitu. Narušení konformace proteinu (např. vlivem glykace) bývá provázeno změnou nebo ztrátou jeho biologické aktivity. Tvorba AGE produktů způsobuje zesítění řetězců proteinů a tím zvýšení jejich molekulové hmotnosti a snížení rozpustnosti. Glykované bílkoviny jsou rovněž schopné tvořit vysokomolekulární agregáty. Následkem změn v prostorovém uspořádání ztrácí proteiny svou flexibilitu, jsou rigidnější. Glykované proteiny jsou vysoce odolné vůči proteolytickému štěpení a mohou se tedy patologicky ukládat v tkáních. Rovněž odolnější vůči denaturaci než nativní proteiny a to zřejmě díky zesítění a/nebo tvorbě disulfidových můstků.
Prvním proteinem, u něhož byla popsána glykace za in vivo podmínek, byl hemoglobin (Hb). Vzhledem k jeho vysoké koncentraci v erytrocytech, jejichž zásobení glukosou je dáno glykemií, se glykovaný Hb stal základem moderní diagnostiky a monitorování DM, protože jeho hladina odráží koncentraci glykemii po celou dobu existence červené krvinky (tj. asi 120 dní) a využívá se k posouzení úspěšnosti léčby/kompenzace diabetu v období 4–8 týdnů před vyšetřením. Výhodou tohoto testu je, že není ovlivněn aktuální hladinou glykémie. Neglykovaný Hb se označuje jako HbA0. V krvi podléhá neenzymové glykaci, při které vznikají 4 deriváty souhrnně označované jako HbA1. Liší se v sacharidu navázaném na N‑terminálním valinu ß-řetězce. Stanovuje se frakce HbA1c s navázanou glukosou (HbA1c má labilní a stabilní formu – labilní je tvořená Schiffovou bazí a stabilní Amadoriho produktem). Glykovaný hemoglobin má jiný náboj než parentní HbA0 a pohybuje se rychleji při elektroforetických či afinitně-chromatografických separacích. U pacientů s DM byly popsány změny ve schopnosti vázat a uvolňovat O2. Glykovaný Hb má vyšší afinitu ke O2, ale obtížněji jej uvolňuje, což může způsobit tkáňovou hypoxii.
Hexosaminová cesta
Jedním z metabolických meziproduktů glykolýzy je fruktosa-6-fosfát, který je zčásti metabolizován na glukosamin-6-fosfát a dále na UDP-GlcNAc, což je substrát pro syntézu proteoglykanů a glykoproteinů. Při protrahované hyperglykemii stoupá produkce aminocukrů, glykosaminoglykanů, proteoglykanů aglykoproteinů, jejichž zvýšený obsah v cévní stěně zvyšuje riziko vzniku angiopatií a atherosklerosy. UDP-GlcNAc je však také ligandem různých transkripčních faktorů (např. Sp1) a ovlivňuje transkripci specifických genů. Aktivace transkripčního faktoru Sp1 vyvolá zvýšení exprese genů pro transformační růstový faktor β1 (TGF-β1) a inhibitor aktivátoru plasminogenu (PAI), které se podílejí na rozvoji fibrosy endotelu a stimulují atherosklerosu.
Aktivace proteinkinasy C
Zvýšená aktivita proteinkinasy C (PKC) je u diabetiků způsobena nadprodukcí DAG a vyvolává aktivaci intracelulárních signálních kaskád, které vyvolávají zvýšení exprese PAI, NF-κB a TGF-β1. Zvýšená množství těchto proteinů vyvolávají změny v endotelu cév a poškození neurovaskulární cirkulace neuronu s následnou ischemizací. Díky aktivaci PKC je snížena exprese endoteliální synthasy NO, která přispívá k vasodilataci, a naopak zvýšena exprese vasokonstrikčně působící látky endotelinu 1. Tyto změny jsou provázeny abnormalitami v proudění krve. Aktivita PKC je rovněž závislá na redoxním stavu buňky (aktivována prooxidačními látkami a inhibována antixodanty), takže lze očekávat zvýšení její aktivity i díky oxidačnímu stresu vyvolanému ostatními patogenetickými mechanismy.
Gestační diabetes mellitus
Jakákoliv intolerance glukosy odpovídající kritériím pro DM, IFG nebo narušení glukosové tolerance, které se poprvé objeví během těhotenství a spontánně odezní během šestinedělí, se označují jako gestační diabetes mellitus (GDM). Tato porucha hospodaření s glukosou se objevuje až u 18 % těhotných. Rizikovými faktory pro rozvoj GDM jsou věk (> 25 let), nadváha či obezita, nedostatek fyzické aktivity před a během těhotenství a nevhodná skladba potravy (např. vysoká konzumace červeného masa).
Patofyziologicky je porucha tolerance glukosy v těhotenství spuštěna působením mateřských a placentárních hormonů a jiných látek (např. lidský placentární laktogen, progesteron, estrogeny, prolaktin, kortisol a leptin), které působí jako antagonisté insulinu a/nebo navozují insulinovou rezistenci. Hladina těchto látek během těhotenství postupně narůstá, proto se GDM rozvíjí až v jeho druhé polovině, kdy je hladina těchto látek výrazně zvýšená. U žen bez dispozice k DM je snížený účinek insulinu kompenzován jeho zvýšenou sekrecí, zatímco u žen s dispozicí k DM je tato kompenzační schopnost omezená. S narůstající insulinovou rezistencí se u nich projeví přechodná porucha tolerance glukosy, která se během šestinedělí opět upraví. Zhruba u 20-30 % žen s GDM se v budoucnu rozvine DM2.
Glukosa prochází placentou vždy po koncentračním spádu, tedy od matky do plodu. U matek s GDM dostává tedy plod vyšší dávky glukosy, což podporuje sekreci insulinu plodem a spouští anabolické procesy podporující růst tukové tkáně, svalů a kostí plodu. Výsledkem je makrosomie a obezita plodu (> 4000 g). Postiženy jsou i jeho vnitřní orgány a dítě je ohroženo poruchou jejich funkce (arytmie, srdeční zástava atd.). Při spontánním porodu takového plodu hrozí větší porodní poranění matky i dítěte a často je nutný instrumentální porod či císařský řez. U makrosomických plodů dochází často ke zpomalení vyzrávání vnitřních orgánů plodu (zejm. nervový a dýchací systém), což s sebou přináší další poporodní komplikace. Další komplikací je novorozenecká hypoglykemie. Díky hyperglykemii a hyperinsulinemii může dojít k poškození funkce β-buněk pankreatu a zvýšit pravděpodobnost vzniku DM2 u těchto dětí v budoucnu.
Jedná se o chronické heterogenní onemocnění provázené hyperglykemií v důsledku dlouhodobého absolutního nebo relativního nedostatku insulinu nebo jeho nedostatečného účinku ve tkáních. Následkem nedostatku insulinu se rozvíjí komplexní metabolická porucha, která zasahuje do metabolismu sacharidů, lipidů a proteinů i do hospodaření s vodou a elektrolyty. V současné době se DM podle American Diabetes Association klasifikuje na čtyři typy: 1) DM 1. typu; 2) DM 2. typu; 3) další specifické typy DM (genetické defekty β-buňky či účinku insulinu, onemocnění exokrinního pankreatu, endokrinopatie – hormony regulace glykemie, lékové a chemické vlivy, infekce, neobvyklé formy imunitně podmíněného DM, jiné genetické syndromy někdy provázející DM); a 4) gestační DM.
Diabetes mellitus 1. typu
DM1 (též označován jako DM1A) je vyvolán autoimunitní destrukcí β-buněk pankreatu, která způsobí absolutní deficit insulinu. Tento typ DM se poprvé manifestuje již v dětství a je nutná celoživotní léčba insulinem. U jednovaječných dvojčat je 30-50% šance, že se tento typ DM objeví u obou sourozenců. Charakteristickými znaky DM1 je rychlý nástup těžké hyperglykemie, úbytek hmotnosti a diabetická ketoacidosa, která může být smrtelná.
K vývoji DM1 je třeba genetická predispozice, která umožní rozpoznání β-buněk jako něčeho cizorodého, a vnější stimul (expozice viru či toxinu). Právě expozice viru/toxinu může u geneticky predisponovaných jedinců spustit destrukci β-buněk T-lymfocyty a makrofágy, která je provázena insulitidou (zánětem) nejprve bez poruchy sekrece insulinu. V krvi pacientů s DM1 jsou přítomné protilátky proti β-buňkám a jejich součástem (protilátky IAA proti insulinu, ICA proti buňkám Langerhansových ostrůvků, GAD proti glutamátdekarboxylase v Langerhansových ostrůvcích), které mají diagnostický význam. V průběhu let jsou postupně ničeny β-buňky a dochází tedy ke snížení tvorby insulinu. Když kapacita pankreatu pro tvorbu insulinu klesne pod prahovou hodnotu (je zničeno 80-90 % β-buněk), objeví se náhle příznaky DM1 a pacient je odkázán na terapii insulinem.
V některých afrických a asijských populacích je popisován idiopatický diabetes s neznámou etiologií. U tohoto typu označovaného také jako DM1B jsou β-buňky zničeny, i když nejsou v krvi přítomné autoprotilátky.
Metabolické změny při DM1
DM1 je typické katabolické onemocnění, které se manifestuje hyperglykemií, glykosurií, ketoacidosou, ketonurií, dyslipidemií, zvýšenou hladinou laktátu, deplecí tukové tkáně a katabolismem proteinů. Patogeneze těchto změn souvisí s nedostatkem insulinu a zvýšenou tvorbou antagonistů insulinu (glukagon, katecholaminy a kortisol). Důsledkem je převaha katabolických reakcí nad reakcemi anabolickými (Obr. 16).
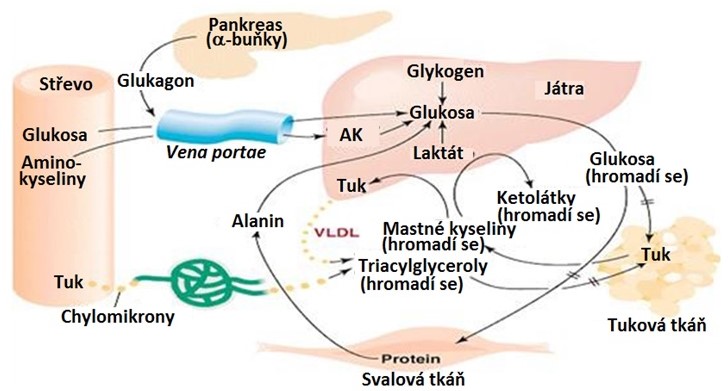
Obr. 16. Hlavní změny v metabolismu živin u neléčeného DM 1. typu (upraveno z Devlin 1997)
Orgány zůstávají i při dostatečném přísunu živin ze střeva v katabolické fázi. Charakteristickou změnou je hyperglykemie, která se může až desetinásobně zvýšit v porovnání s normálními hladinami (~ 5 mmol/l). Příčinou vzniku hyperglykemie je jednak snížení příjmu glukosy do insulin-dependentních tkání (tuková tkáň, srdeční a kosterní svalstvo), protože vlivem nedostatku insulinu nejsou tyto tkáně schopné vystavit na svých membránách transportér GLUT4. Dalším důvodem je inhibice glykolýzy (inhibice klíčových enzymů a pyruvátdehydrogenasy) a aktivace glukoneogeneze (aktivace klíčových enzymů), glykogenolýzy (inhibice glykogensynthasy a aktivace glykogenfosforylasy) a uvolnění glukosy z jater do krve. Díky inhibici glykolýzy a syntézy glykogenu nemůže být glukosa vytvořená v glukoneogenezi v játrech využita a je i přes vysokou koncentraci v extracelulární tekutině transportována do krve. Pokud hladina glukosy v krvi překročí transportní kapacitu ledvin, pak se glukosa objevuje v moči (glykosurie), což se projeví osmotickou diurézou a rychle se rozvíjející dehydratací, pokud není adekvátně zvýšen příjem tekutin.
Díky nízkému poměru insulin/glukagon je v tukové tkáni inhibována lipoproteinová lipasa a aktivována hormon-sensitivní lipasa, což vede k nekontrolované lipolýze a do krve se dostává velké množství mastných kyselin. Jejich oxidace v řadě tkání inhibuje oxidaci glukosy (tzv. Randleův cyklus), protože vznikající meziprodukty (citrát, acetyl-CoA, NADH) přímo inhibují metabolické dráhy oxidace glukosy (pyruvátdehydrogenasa, fosfofruktokinasa), což vyvolá nahromadění glukosa-6-fosfátu, který inhibuje hexokinasu a následně dojde k omezení schopnosti svalových buněk přijímat a zpracovávat glukosu. Játra zpracovávají mastné kyseliny částečně na triacylglyceroly, které jsou ve formě VLDL secernovány do krve. Jejich zpracování však kvůli inhibici lipoproteinové lipasy vázne a rozvíjí se hypertriacylglycerolemie. Při β‑oxidaci mastných kyselin vzniká velké množství acetyl-CoA, který nemůže všechen vstoupit do citrátového cyklu (nedostatek oxalacetátu kvůli probíhající glukoneogenezi), je jeho část převedena do syntézy ketolátek a cholesterolu. Ketolátky se vylučují především močí, část acetonu se eliminuje plícemi. Při vysokých koncentracích ketolátek dojde k překročení kapacity ledvin a ketolátky se objevují v moči (ketonurie). Zároveň dojde k poruše acidobazické rovnováhy, protože ketolátky mají povahu středně silných kyselin a působí tedy při vysokých koncentracích jako pufry, takže mohou vyčerpat alkalickou zásobu a pak dojde k rozvoji ketoacidosy. Nahromadění kyselin a vznikající acidosa nutí pacienty k hlubokému dýchání (Kussmaulovo dýchání).
Celkový obrat proteinů u neléčeného DM1 v důsledku zvýšeného proteokatabolismu a snížené syntézy proteinů stoupá a zvyšují se ztráty dusíku močí. Rozkladem proteinů je postiženo hlavně kosterní svalstvo. Jedním z hlavních energetických substrátů pro sval se stávají aminokyseliny s větveným řetězcem (stoupá aktivita komplexu dehydrogenas 2‑oxokyselin s větveným řetězcem). S aktivací proteolýzy ve svalech a degradací aminokyselin s větveným řetězcem je spojena zvýšená syntéza alaninu a glutaminu. Většina aminokyselin uvolněných svalovými buňkami do krve je využita ve viscerálních tkáních jako energetický zdroj a jako substrát pro syntézu glukosy.
Diabetes mellitus 2. typu
Jedná se o nejčastější formu DM (až 90 % z celkového počtu diabetiků) a manifestuje se obvykle u starších a obézních osob. V posledních letech se však významně zvýšil výskyt DM2 u obézních adolescentů. V tomto případě je nedostatek insulinu relativní. DM2 se rozvíjí pomalu, často po řadu let. Genetické vlivy jsou zde silnější než u DM1, jednovaječná dvojčata mají až 72% pravděpodobnost, že budou obě postižená touto nemocí. DM2 je charakterizován hyperglykemií, glykosurií a dyslipidemií podobného charteru jako DM1. Hladina ketolátek je však obvykle normální a nedochází ani k rozvoji proteokatabolismu. Většina metabolických odchylek pozorovaných u tohoto typu DM je vyvolána rezistencí tkání k insulinu.
Insulinová rezistence je stav, při němž fyziologické množství insulinu vyvolá subnormální biologickou odpověď. Je způsobena sníženým počtem receptorů nebo jejich necitlivostí k insulinu. Na vzniku insulinové rezistence se podílejí vlivy vrozené (např. mutace insulinového receptoru, transportérů pro glukosu či signální proteiny) a vlivy získané (např. obezita, vysoká koncentrace volných mastných kyselin, přejídání, stáří, medikace). Genetické defekty mohou být pre-receptorové (např. abnormální insulin, protilátky proti insulinu), receptorové (např. menší počet receptorů, omezená schopnost vázat insulin, mutace receptoru, jeho zablokování protilátkami) či post-receptorové (např. poruchy v přenosu signálu, mutace GLUT4). Nejčastější příčinou však je obezita, do jejíhož průběhu se významně zapojují hormony produkované tukovou tkání, tzv. adipokiny (leptin, adiponektin, rezistin, visfatin a další). Pokud jsou β-buňky schopné produkovat insulin, pak dochází při zvýšených požadavcích organismu na insulin ke zvýšení jeho produkce (hyperinsulinemie).
Metabolické změny při DM2
U DM2 je insulin přítomen, ale tkáně jsou vůči němu méně vnímavé. Charakteristickým znakem DM2 je hyperglykemie, která vzniká v důsledku sníženého příjmu glukosy do periferních tkání (hlavně buněk kosterního svalstva) a zvýšené jaterní glukoneogeneze. Dalším průvodním znakem tohoto onemocnění je hypertriacylglycerolemie, která vzniká v důsledku rychlé de novo syntézy mastných kyselin a VLDL v játrech spíše než kvůli zvýšené dodávce mastných kyselin z tukové tkáně (DM1). Nekontrolované lipolýza totiž není u tohoto typu DM přítomná a díky přítomnosti insulinu nevzniká ani ketoacidosa (Obr. 17).
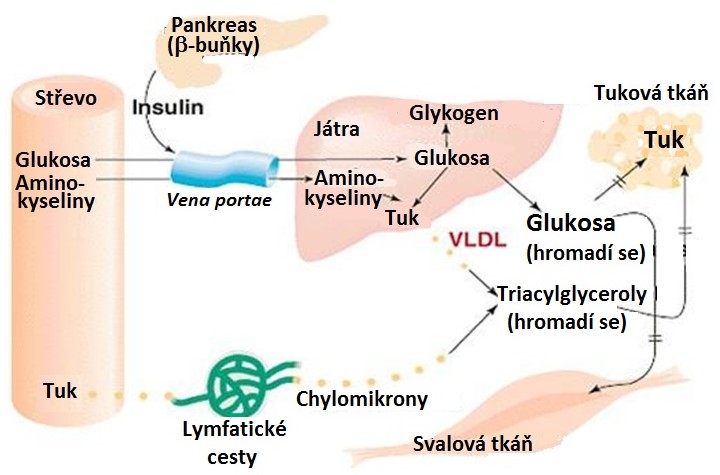
Obr. 17. Hlavní změny v metabolismu živin u neléčeného DM 2. typu (upraveno z Devlin 1997)
Na patogenezi insulinové rezistence se podílejí i volné mastné kyseliny (VMK), protože obézní jedinci a pacienti s DM2 mají vyšší hladiny VMK než zdraví jedinci. Při zvýšené nabídce VMK dochází v tkáních k jejich přednostní oxidaci a k inhibici drah oxidujících glukosu (Randleův cyklus). Při nízké aktivitě svalové hexokinasy se omezuje schopnost svalové buňky přijímat a zpracovávat glukosu a rozvíjí se insulinové rezistence na úrovni svalové buňky. Kromě toho dochází ve svalové buňce k nahromadění meziproduktů oxidace mastných kyselin (acyl-CoA s dlouhým řetězcem), které inhibují enzymy syntézy glykogenu a utilizace glukosy. Tyto meziprodukty mohou přeměněny též na diacylglycerol (DAG), významnou signální molekulu, který aktivací proteinkinasy C vyvolává abnormální fosforylaci IRS a tím inhibici signalizační dráhy insulinového receptoru a tím i snížení transportu glukosy do svalové buňky. V játrech je v důsledku zvýšené nabídky VMK aktivována glukoneogeneze bez současné inhibice glykogenolýzy (autoregulační mechanismus u zdravých osob) a játra tak uvolňují glukosu do krve v situaci, kdy nejsou svalové buňky schopné tento nadbytek glukosy přijmout. Pokud má být za této situace udržena normální hladina glukosy, musí se zvýšit tvorba insulinu v pankreatu. Při krátkodobém (do 6 hodin) vystavení β-buněk pankreatu působení zvýšených hladin VMK dochází ke zvýšení glukosou-stimulovaného výdeje insulinu. Při dlouhodobé expozici VMK však dochází ke snížení produkce insulinu (asi díky navození oxidačního stresu v β-buňkách), tak ke snížení periferní vnímavosti tkání k insulinu. V této situaci je porušena regulace glykemie, rozvíjí se porucha glukosové tolerance a DM2.
Komplikace diabetu mellitu
U obou typů diabetu dochází k řadě komplikací, které vznikají v návaznosti na popsané metabolické změny. Tyto komplikace můžeme rozdělit na akutní a chronické. Mezi akutní komplikace DM patří hypoglykemické kóma, hyperglykemické diabetické kóma s ketoacidosou, hyperosmolární kóma bez ketoacidosy a laktacidotické kóma. Chronické komplikace DM, které můžeme rozdělit na mikro- a makrovaskulární komplikace, jsou představovány degenerativními změnami krevních a nervového systému.
Akutní komplikace diabetes mellitus
Při poklesu glykemie pod 3,3 mmol/l hovoříme o hypoglykemii. Hypoglykemie se vyskytuje častěji u pacientů s DM1. Zhruba jedna třetina diabetiků léčených insulinem prodělá alespoň jednou v životě hypoglykemické kóma a 2-4 % pacientů s DM1 na hypoglykemii zemřou. Příčinou vzniku hypoglykemie je nejčastěji podání příliš velké dávky insulinu, nedostatečný příjem potravy nebo nadměrná či nepředpokládaná fyzická zátěž. Při hypoglykemii dochází nejprve k aktivaci kontraregulačních mechanismů, která se projeví neklidem, třesem, pocením, zčervenáním, tachykardií a pocitem hladu. Při pokračující hypoglykemii se rozvíjí symptomy porušené funkce CNS (neuroglykopenie) – snížení psychomotorických a intelektuálních funkcí, které jsou při dalším poklesu glykemie provázeny poruchami vědomí až kómatem. Akutní léčba sestává z p.o. podání 10-20 g volných sacharidů (tj. 2-4 kostky cukru), které lze po 5-10 minutách opakovat. U pacientů s poruchou vědomí se podává 20% glukosa i.v. a glukagon i.m.
Diabetická ketoacidosa je nejčastější příčinou úmrtí diabetiků do 20 let věku, mortalita je kolem 5 %. Častěji se vyskytuje u pacientů s DM1. Je provázena hyperglykemií (≥ 15 mmol/l), která je příčinou vystupňované osmotické diurézy vyvolávající dehydrataci pacienta. Dehydratace vede k hyperosmolaritě, která může vést až k poruše vědomí a kómatu. Metabolická acidosa se rozvíjí díky vystupňované syntéze ketolátek v játrech (ketonemie > 5 mmol/l) a je často provázena hyperventilací (Kussmaulovo dýchání). Příčinou vzniku tohoto stavu je absolutní nebo relativní nedostatek insulinu či nadbytek kontraregulačních hormonů. Při léčbě se doplňují chybějící tekutiny, insulin, minerály, upravuje se vnitřní prostředí a intenzivně se léčí vyvolávající příčina (např. podání antibiotik u infekcí, koronární intervence u infarktu myokardu).
Hyperosmolární syndrom bez ketoacidosy je akutní komplikací především DM2 s vysokou mortalitou (15-30 %). Projevuje se extrémní hyperglykemií (> 35–50 mmol/l) s těžkou dehydratací a hyperosmolaritou plasmy, rizikem vzniku prerenální renální insuficience různého stupně a poruchami vědomí. Ketoacidóza není přítomná. Mezi nejčastější příčiny vzniku patří kardiovaskulární příhody, rozsáhlejší infekce či nepřiměřená terapie některými léčivy (diuretika, kortikoidy, β-blokátory), které znemožní nemocnému dostatečný příjem tekutin při osmotické diuréze ze stoupající glykemie. V patogenezi se uplatňuje relativní nedostatek insulinu s nadbytkem kontraregulačních hormonů. Léčebná opatření zahrnují vždy hospitalizaci na jednotce intenzivní péče, i.v. podání tekutin, insulinu, korekce mineralogramu, komplexní léčba šokového stavu a vyvolávající příčiny (antibiotika, antikoagulační léčba atd.).
Laktátová acidosa je metabolická acidosa (pH < 7,35) se zvýšenou hladinou laktátu v krvi (> 6 mmol/l), která pacienta ohrožuje na životě a má značnou mortalitu (až 50 %). Jedná se o vzácnou komplikaci pacientů s DM2 léčených biguanidy (zejm. fenformin a buformin). Objevuje se u nemocných, u nichž nebyly dodrženy kontraindikace léčby biguanidy. Projevy jsou často nespecifické, a pokud není laktátová acidosa včas rozpoznána, rozvíjí se těžká metabolická acidosa se vzestupem laktátu a se závažnými změnami celkového stavu pacienta, které často končí smrtí dotyčného. Léčba probíhá na jednotce intenzivní péče za použití hemopurifikačních metod.
Chronické komplikace diabetes mellitus
Při dlouhodobém působení hyperglykemie dochází po letech až desetiletích k nevratným změnám v organismu, které označujeme jako chronické komplikace DM. Ty můžeme rozdělit na makrovaskulární a mikrovaskulární a hovoříme o diabetické mikro- a makroangiopatii. Diabetická makroangiopatie (ischemická choroba srdeční, cévní mozkové příhody, ischemická choroba dolních končetin) je souhrnné označení pro aterosklerotické projevy na velkých tepnách diabetiků. Diabetická atherosklerosa je charakteristická časnějším vznikem a rychlejší progresí. Mezi klasické mikrovaskulární komplikace patří diabetická nefropatie, retinopatie a neuropatie.
V rozvoji mikro- i makrovaskulárních komplikací hraje významnou úlohu oxidační stres. Z dlouhodobých výzkumů chronických komplikací diabetu vyplývá, že hlavní příčinou metabolických abnormalit pozorovaných u DM je nadprodukce reaktivních forem kyslíku (ROS) mitochondriemi. Nadbytek ROS amplifikuje další tvorbu ROS aktivací NADPH‑oxidas a endoteliální synthasy NO. Stabilní ROS difundují do jádra, kde poškozují DNA, čímž je aktivována poly(ADP-ribosa)polymerasa (PARP). Ribosylace glyceraldehyd-3-fosfátdehydrogenasy (GAPDH) katalyzovaná PARP snižuje aktivitu GAPDH, což vyvolá hromadění časných meziproduktů glykolýzy, které se následně přesouvají do čtyř patogenních signálních drah. Díky inhibici GAPDH stoupá koncentrace glyceraldehyd-3-fosfátu (GAP), který je metabolizován na methylglyoxal, jeden z velmi reaktivních meziproduktů neenzymové glykace (viz. dále). Isomerizací GAP vzniká dihydroxyacetonfosfát, který může být metabolizován na DAG, významný aktivátor proteinkinasy C. Fruktosa-6-fosfát může být přesměrován do hexosaminové dráhy, kde z něj vzniká UDP-N-acetylglukosamin (UDP‑GlcNAc), který působí jako ligand některých transkripčních faktorů a mění transkripci specifických genů. Nadbytek glukosy jev polyolové cestě přeměněn aldosareduktasou na cukerný alkohol sorbitol, který díky zvýšení osmotického tlaku v buňce vyvolává poškození oční čočky, nervů a glomerulů ledvin. Cesty tkáňového poškození vyvolané hyperglykemií jsou shrnuty v Obr. 18.
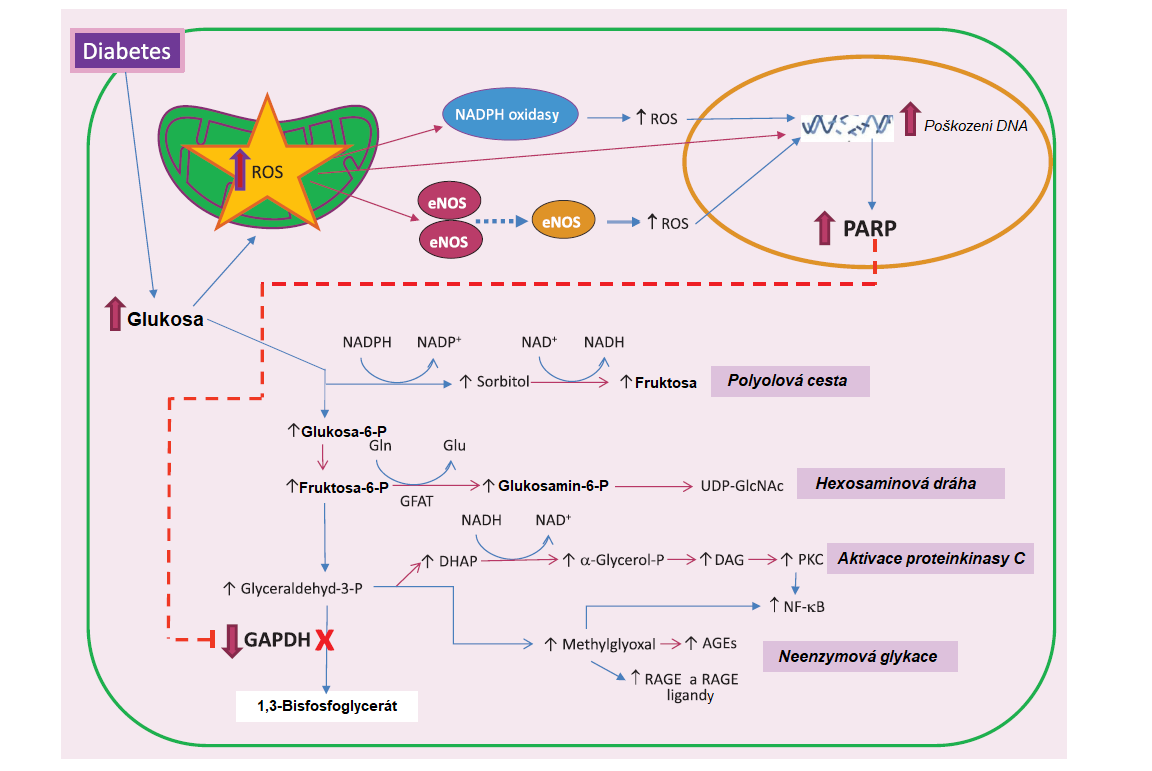
Obr. 18. Čtyři patogenní mechanismy indukované hyperglykemií jsou aktivovány nadprodukcí ROS (upraveno z Shah a Brownlee 2016). AGEs, koncové produkty pokročilé glykace; DAG, diacylglycerol; DHAP, dihydroxyacetonfosfát; eNOS, endoteliální synthasa oxidu dusnatého; GAPDH, glyceraldehyd-3-fosfátdehydrogenasa; GFAT, glutamin:fruktosa-6-fosfát-amidotransferasa; Glu, glutamát; Gln, glutamin; NF-κB, nukleární faktoru κB; PARP, poly(ADP-ribosa)polymeráza; PKC, proteinkinasa C; RAGE, receptor pro AGE; ROS, reaktivní formy kyslíku; UDP-GlcNAc, UDP-N-acetylglukosamin
Polyolová cesta
Glukosa se v buňkách, které obsahují aldosareduktasu (např. oční čočka, neurony, glomeruly), redukuje na cukerný alkohol sorbitol (Obr. 19). Tato dráha je aktivována hyperglykemií, protože KM aldosareduktasy pro glukosu je vysoké (70 mmol/l). Sorbitol nemůže díky své silně hydrofilní povaze přecházet přes buněčné membrány, v buňce se hromadí a osmoticky přitahuje vodu, což vyvolává osmotický edém buňky. V případě oční čočky vyvolává edém denaturaci proteinů krystalínů, které vytvářejí ložiska rozptylující světlo, a tak vzniká zákal čočky (katarakta). Reakce katalyzovaná aldosareduktasou spotřebovává NADPH, který je nezbytný např. k udržení hladiny redukovaného glutathionu (významný antioxidant) v buňce, takže jsou buňky méně chráněné před oxidačním stresem. Při zvýšení poměru NADP+/NADPH se rozvíjí diabetická pseudohypoxie. Aktivita polyolové cesty může rovněž přispívat k depleci NAD+, protože sorbitol je sorbitoldehydrogenasou (SDH) oxidován za účasti NAD+ na fruktosu. Díky spotřebě NAD+ kompetuje SDH s GAPDH, díky čemuž se v buňce hromadí triosafosfáty, které se mohou stát prekurzory tvorby AGEs. Vznikající fruktosa pak může rovněž přispět k tvorbě AGEs, protože je nejprve fosforylována na fruktosa-3-fosfát, který je degradován na 3-deoxyglukoson. Oba tyto produkty jsou silnými glykačními činidly a podporují vznik AGE produktů.
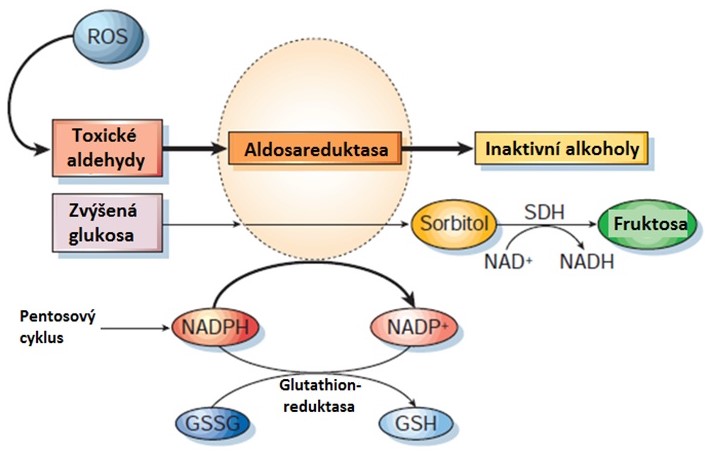
Obr. 19. Aldosareduktasa a polyolová cesta (upraveno z Brownlee 2001). GSH, redukovaný glutathion, GSSG, oxidovaný glutathion
Neenzymová glykace (tvorba AGE produktů)
Neenzymová glykace (též Maillardova reakce) patří mezi posttranslační modifikace proteinů. Probíhá na volných aminoskupinách proteinů, fosfolipidů a nukleových kyselin, kde dochází k navázání molekul redukujících sacharidů. Ty jsou sledem následných reakcí přeměněny až na koncové AGE produkty. Glykace probíhá ve všech organismech kontinuálně během celého života a významnou měrou se podílí na změnách provázejících stárnutí organismu. Všichni jsme tedy v průběhu života vystaveni působení glukosy, a čím vyšší je její koncentrace v krvi, tím závažnější jsou následky. Neenzymová glykace se významně podílí na patogenezi a progresi celé řady onemocnění včetně DM.
Neenzymová glykace je komplexní proces (Obr. 20), který je v časných fázích závislý na koncentraci glukosy. Probíhá pomalu, týdny až měsíce, a je zahájen kondenzací redukujícího cukru nebo podobné sloučeniny (např. askorbátu) s volnou aminoskupinou proteinu, lipidu nebo nukleové kyseliny. V případě glukosy vzniká v rozmezí několika hodin nestabilní aldimin známý jako Schiffova báze, který přesmykem poskytuje relativně stabilní ketoamin, tzv. Amadoriho produkt (např. fruktosamin). Tvorba Amadoriho produktu je do určité míry vratným procesem, avšak rovnováha reakce je výrazně posunuta ve prospěch jeho vzniku. Klasickým přesmykem je Amadoriho produkt postupně přeměněn na AGEs a to buď za neúčasti, nebo při účasti kyslíku (glykoxidace). Schiffova báze i Amadoriho produkt jsou ve střední fázi glykační reakce degradovány oxidačními a dehydratačními reakcemi na řadu α‑dikarbonylových sloučenin (např. glyoxal, methylglyoxal a 3-deoxyglukoson). Rovněž autooxidace glukosy katalyzovaná ionty přechodných kovů je zdrojem dikarbonylových sloučenin. Ty reagují s proteiny ochotněji než parentní monosacharid a vytvářejí různé zkřížené vazby a AGE produkty. Kromě dikarbonylových sloučenin vznikají rovněž ROS (např. peroxid vodíku), které iniciují další oxidační kroky a podílejí se tak na vzniku AGE produktů. Během týdnů až měsíců vznikají v pozdní fázi koncové produkty pokročilé glykace. Jejich vznik je urychlen přítomností oxidačního a karbonylového stresu.
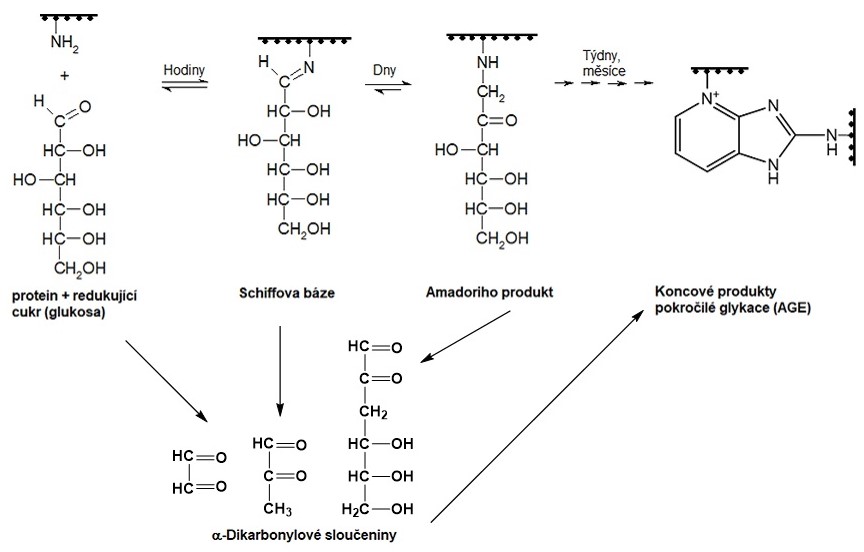
Obr. 20. Průběh glykační reakce
AGE produkty tvoří heterogenní skupinu látek, pro něž je charakteristická žlutohnědá pigmentace, fluorescence, schopnost vzájemného zesítění (crosslinking) a reakce s receptorem RAGE, který je specifický pro AGEs. Existují ovšem i AGE nefluoreskující a AGE, které schopnost zesítění nemají. Struktura řady AGE není známa, ale některé z nich byly popsány jako modelové AGE a další jsou postupně charakterizovány. Zřejmě nejvýznamnějším AGE produktem schopným tvořit zesítění je pro člověka glukosapan, jehož tkáňová koncentrace až tisícinásobně převyšuje koncentrace dalších AGE produktů schopných tvořit zesítění. Kromě endogenní tvorby AGEs pocházejí také z exogenních zdrojů (např. potrava, tabákový kouř).
Prekursory AGE produktů mohou být v organismu detoxifikovány pomocí specifických reduktas (např. glyoxalasou). AGE-modifikované proteiny jsou vychytány makrofágy díky scavengerovým receptorům na jejich povrchu. Tyto proteiny jsou intracelulárně degradovány a uvolněné malé rozpustné AGE-peptidy a AGE-adukty jsou vyloučeny ledvinami. Snížená funkce ledvin či poškození funkce jater může vést ke zvýšení jejich plazmatických koncentrací a k hromadění v tkáních, které má řadu toxických účinků. AGE mohou přímo poškodit strukturu mezibuněčné hmoty, změnit její fyzikální a chemické vlastnosti, metabolismus a způsobit abnormální zesítění, blokovat vasodilatační působení oxidu dusnatého, indukovat lipoperoxidaci nebo působit přes specifické receptory. Těch bylo popsáno několik a nejvýznamnějším z nich je RAGE. Jeho aktivace vede ke spuštění signální kaskády zahrnující aktivaci NF-κB, která je spojena s oxidačním stresem. Následuje stimulace transkripce genů pro prozánětlivé cytokiny, růstové faktory a adhezivní molekuly.
Glykace proteinů s sebou přináší celou řadu následků pro jejich strukturu a aktivitu. Narušení konformace proteinu (např. vlivem glykace) bývá provázeno změnou nebo ztrátou jeho biologické aktivity. Tvorba AGE produktů způsobuje zesítění řetězců proteinů a tím zvýšení jejich molekulové hmotnosti a snížení rozpustnosti. Glykované bílkoviny jsou rovněž schopné tvořit vysokomolekulární agregáty. Následkem změn v prostorovém uspořádání ztrácí proteiny svou flexibilitu, jsou rigidnější. Glykované proteiny jsou vysoce odolné vůči proteolytickému štěpení a mohou se tedy patologicky ukládat v tkáních. Rovněž odolnější vůči denaturaci než nativní proteiny a to zřejmě díky zesítění a/nebo tvorbě disulfidových můstků.
Prvním proteinem, u něhož byla popsána glykace za in vivo podmínek, byl hemoglobin (Hb). Vzhledem k jeho vysoké koncentraci v erytrocytech, jejichž zásobení glukosou je dáno glykemií, se glykovaný Hb stal základem moderní diagnostiky a monitorování DM, protože jeho hladina odráží koncentraci glykemii po celou dobu existence červené krvinky (tj. asi 120 dní) a využívá se k posouzení úspěšnosti léčby/kompenzace diabetu v období 4–8 týdnů před vyšetřením. Výhodou tohoto testu je, že není ovlivněn aktuální hladinou glykémie. Neglykovaný Hb se označuje jako HbA0. V krvi podléhá neenzymové glykaci, při které vznikají 4 deriváty souhrnně označované jako HbA1. Liší se v sacharidu navázaném na N‑terminálním valinu ß-řetězce. Stanovuje se frakce HbA1c s navázanou glukosou (HbA1c má labilní a stabilní formu – labilní je tvořená Schiffovou bazí a stabilní Amadoriho produktem). Glykovaný hemoglobin má jiný náboj než parentní HbA0 a pohybuje se rychleji při elektroforetických či afinitně-chromatografických separacích. U pacientů s DM byly popsány změny ve schopnosti vázat a uvolňovat O2. Glykovaný Hb má vyšší afinitu ke O2, ale obtížněji jej uvolňuje, což může způsobit tkáňovou hypoxii.
Hexosaminová cesta
Jedním z metabolických meziproduktů glykolýzy je fruktosa-6-fosfát, který je zčásti metabolizován na glukosamin-6-fosfát a dále na UDP-GlcNAc, což je substrát pro syntézu proteoglykanů a glykoproteinů. Při protrahované hyperglykemii stoupá produkce aminocukrů, glykosaminoglykanů, proteoglykanů aglykoproteinů, jejichž zvýšený obsah v cévní stěně zvyšuje riziko vzniku angiopatií a atherosklerosy. UDP-GlcNAc je však také ligandem různých transkripčních faktorů (např. Sp1) a ovlivňuje transkripci specifických genů. Aktivace transkripčního faktoru Sp1 vyvolá zvýšení exprese genů pro transformační růstový faktor β1 (TGF-β1) a inhibitor aktivátoru plasminogenu (PAI), které se podílejí na rozvoji fibrosy endotelu a stimulují atherosklerosu.
Aktivace proteinkinasy C
Zvýšená aktivita proteinkinasy C (PKC) je u diabetiků způsobena nadprodukcí DAG a vyvolává aktivaci intracelulárních signálních kaskád, které vyvolávají zvýšení exprese PAI, NF-κB a TGF-β1. Zvýšená množství těchto proteinů vyvolávají změny v endotelu cév a poškození neurovaskulární cirkulace neuronu s následnou ischemizací. Díky aktivaci PKC je snížena exprese endoteliální synthasy NO, která přispívá k vasodilataci, a naopak zvýšena exprese vasokonstrikčně působící látky endotelinu 1. Tyto změny jsou provázeny abnormalitami v proudění krve. Aktivita PKC je rovněž závislá na redoxním stavu buňky (aktivována prooxidačními látkami a inhibována antixodanty), takže lze očekávat zvýšení její aktivity i díky oxidačnímu stresu vyvolanému ostatními patogenetickými mechanismy.
Gestační diabetes mellitus
Jakákoliv intolerance glukosy odpovídající kritériím pro DM, IFG nebo narušení glukosové tolerance, které se poprvé objeví během těhotenství a spontánně odezní během šestinedělí, se označují jako gestační diabetes mellitus (GDM). Tato porucha hospodaření s glukosou se objevuje až u 18 % těhotných. Rizikovými faktory pro rozvoj GDM jsou věk (> 25 let), nadváha či obezita, nedostatek fyzické aktivity před a během těhotenství a nevhodná skladba potravy (např. vysoká konzumace červeného masa).
Patofyziologicky je porucha tolerance glukosy v těhotenství spuštěna působením mateřských a placentárních hormonů a jiných látek (např. lidský placentární laktogen, progesteron, estrogeny, prolaktin, kortisol a leptin), které působí jako antagonisté insulinu a/nebo navozují insulinovou rezistenci. Hladina těchto látek během těhotenství postupně narůstá, proto se GDM rozvíjí až v jeho druhé polovině, kdy je hladina těchto látek výrazně zvýšená. U žen bez dispozice k DM je snížený účinek insulinu kompenzován jeho zvýšenou sekrecí, zatímco u žen s dispozicí k DM je tato kompenzační schopnost omezená. S narůstající insulinovou rezistencí se u nich projeví přechodná porucha tolerance glukosy, která se během šestinedělí opět upraví. Zhruba u 20-30 % žen s GDM se v budoucnu rozvine DM2.
Glukosa prochází placentou vždy po koncentračním spádu, tedy od matky do plodu. U matek s GDM dostává tedy plod vyšší dávky glukosy, což podporuje sekreci insulinu plodem a spouští anabolické procesy podporující růst tukové tkáně, svalů a kostí plodu. Výsledkem je makrosomie a obezita plodu (> 4000 g). Postiženy jsou i jeho vnitřní orgány a dítě je ohroženo poruchou jejich funkce (arytmie, srdeční zástava atd.). Při spontánním porodu takového plodu hrozí větší porodní poranění matky i dítěte a často je nutný instrumentální porod či císařský řez. U makrosomických plodů dochází často ke zpomalení vyzrávání vnitřních orgánů plodu (zejm. nervový a dýchací systém), což s sebou přináší další poporodní komplikace. Další komplikací je novorozenecká hypoglykemie. Díky hyperglykemii a hyperinsulinemii může dojít k poškození funkce β-buněk pankreatu a zvýšit pravděpodobnost vzniku DM2 u těchto dětí v budoucnu.