Sled biochemických a morfologických změn při akutním ischemickém a hypoxickém poškození
Reverzibilní poškození
Hypoxie nejprve ovlivní aerobní respiraci buňky, tedy oxidační fosforylaci v mitochondriích. Jak klesá tenze O2, tak se snižuje i oxidační fosforylace a tím i tvorba ATP. Pokles produkce ATP postihuje celou řadu buněčných funkcí a systémů. Kardiomyocyt ztrácí schopnost kontraktility už po 60 s po vzniku koronární okluze, ale to ještě neznamená jeho zánik. Snížení koncentrace ATP provázené vzestupem koncentrace AMP aktivuje fosfofruktokinasu a glykogenfosforylasu, což vede ke zvýšení aktivity anaerobní glykolýzy a glykogenolýzy – náhradních zdrojů ATP. Zásoby glykogenu i kreatinfosfátu jsou však rychle vyčerpány. Anaerobní glykolýzu provází hromadění laktátu, hydrolýzu makroergních fosfátových vazeb zase hromadění anorganického fosfátu, což oboje snižuje intracelulární pH. Následkem změny pH dochází v této časné fázi buněčného poškození k zaškrcování chromatinu.
Deplece ATP je zodpovědná za akutní buněčný edém, který je způsoben poruchou regulace buněčného objemu plasmatickou membránou. Nedostatek ATP vede k selhání membránového transportu iontů (Na/K-ATPasa), dochází k volné difuzi Na+ po koncentračním spádu do buňky a K+ z buňky. To je provázeno isoosmotickým nárůstem vody v buňce, což způsobí buněčný edém a následně dilataci ER. Další příčinou hromadění vody v buňce je nárůst katabolitů (laktát, anorganický fosfát, purinové nukleotidy z NADH), které zvyšují intracelulární osmotickou nálož. Dochází k oddělení ribosomů od ER a disociaci polysomů na monosomy.
Při pokračující hypoxii dochází k prohloubení membránové permeability a dalšímu snížení aktivity mitochondrií. V této fázi bobtnají mitochondrie, celá buňka je hyperhydratovaná díky zvýšenému obsahu Na+, který přitahuje vodu. Vázne proteosyntéza, buňky ztrácejí ultrastrukturní rysy (např. mikrovilli), v cytoplasmě vznikají myelinové obrazce jako důsledek degenerace buněčné membrány (disociace fosfolipodů odhalí fosfatidy). Až do určitého stavu jsou všechny tyto změny vratné, pokud se obnoví přísun kyslíku (Obr. 8).
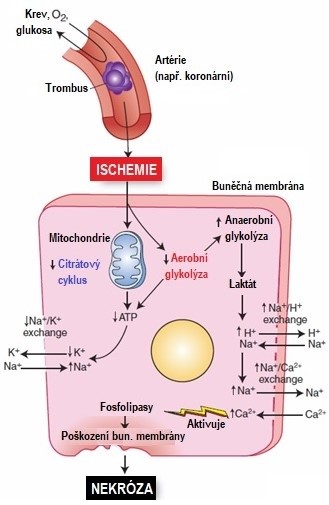
Obr. 8. Biochemické změny při ischemickém poškození buňky (upraveno ze Strayer a Rubin 2011).
Ireverzibilní poškození
Pokud ischemie přetrvává, stává se poškození buňky nevratným. Dochází k výrazné vakuolizaci mitochondrií, značnému poškození membrán a k bobtnání lyzosomů. V kardiomyocytech se tyto známky poškození objevují po 30-40 minutách ischemie. Následně dochází k masivnímu přesunu Ca2+ do cytosolu (hlavně v ischemických oblastech myokardu, kde již došlo k reperfuzi). Pokračuje únik proteinů, enzymů, koenzymů a RNA hyperpermeabilní membránou. Buňky ztrácejí metabolity nutné k syntéze ATP, což způsobí úplnou depleci makroergních fosfátů. V tomto stadiu praská lyzosomální membrána a hydrolytické enzymy (proteasy, fosfatasy, glykosidasy, RNAsy, DNAsy, kathepsiny) se dostávají do cytoplasmy. Kyselé prostředí cytoplasmy tyto enzymy aktivuje, a tak dochází k natrávení buněčných komponent. V konečné fázi je mrtvá buňka vyplněna fosfolipidovou hmotou, čemuž se říká myelinová degenerace.
Ireverzibilní poškození charakterizují dva jevy: neschopnost nápravy mitochondriální dysfunkce po reperfuzi/reoxygenaci a rozvoj hluboké poruchy membránových funkcí. Na poškození membrány se podílí progresivní ztráta fosfolipidů (aktivita endogenních fosfolipas, pokles reacylačních procesů a de novo biosyntézy fosfolipidů), abnormity cytoskeletu (svrašťování a ruptury membrány v důsledku štěpení filament cytoskeletu proteasami aktivovanými Ca2+), reaktivní formy kyslíku (reperfuzní paradox), rozkladné produkty lipidů (detergentní účinek rozkladných produktů – neesterifikovaných MK, acylkarnitinu, lysofosfolipidů) a ztráta intracelulárních aminokyselin.