Hemoglobinopatie
Hemoglobinopatie jsou vrozené poruchy syntézy proteinové části molekuly hemoglobinu, u nichž se na základě genetického defektu tvoří některý z globinových řetězců s abnormální strukturou nebo ve změněném množství ve srovnání se zdravým jedincem.
Jde o jedny z nejčastějších dědičných onemocnění ve světě. Zvýšený výskyt některých hemoglobinopatií je charakteristický pro oblasti s výskytem malárie, neboť heterozygotní formy těchto onemocnění poskytují určitou ochranu proti malarické infekci.
Vzhledem ke genetickému zápisu struktury jednotlivých řetězců (viz obr. 39 ilustrující lokalizaci genů pro jednotlivé řetězce na chromosomech) postihne mutace β genu u heterozygotů 50 % řetězců hemoglobinu, kdežto mutace α genu postihuje jen 25 % molekul, ale projevuje se již před narozením.
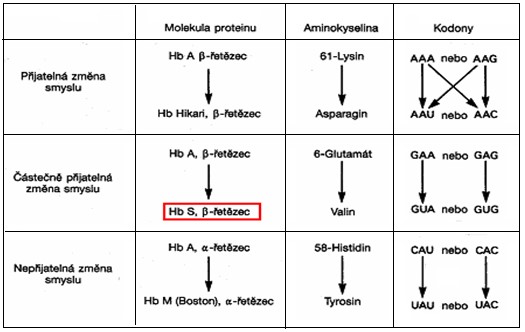
Tabulka 6. Příklady hemoglobinopatií s různou klinickou závažností změny (Murray et al. 1998)
Srpkovitá anémie
Srpkovitá anémie (srpkovitost; sicle cell disease; falciformní anémie; drepanocytóza) je autosomálně recesivní dědičné onemocnění, které se projevuje změnou tvaru červených krvinek. Postižené erytrocyty se jeví v mikroskopu jako protažené srpky. Tato změna tvaru je způsobena mutací genu pro hemoglobin, při níž je na 6. pozici od N-konce v β-řetězci valin místo glutamové kyseliny (vzniká hemoglobin označovaný jako „HbS“) (Obr. 47).
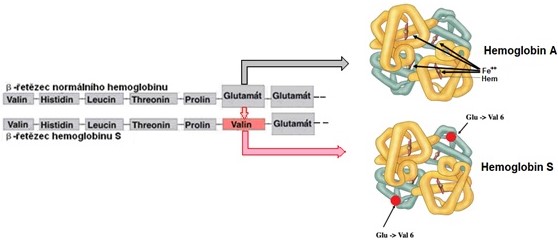
Obr. 47. Záměna glutamátu za valin v β-řetězci hemoglobinu u srpkovité anémie (Přispěvatelé StudyBlue 2016)
Nositel jednoho zdravého genu a jednoho genu HbS (heterozygot) je přenašečem („sickle cell trait“). Přenašeč srpkovité anémie produkuje okolo 20-40 % hemoglobinu S a také dostatečné množství hemoglobinu A (přibližně 60 %) a prakticky nemá žádné zdravotní obtíže. Poškozený gen pak může být předán do další generace. Srpkovitá anémie jako choroba se projevuje u homozygotů.
Zatímco kyselina glutamová je hydrofilní a podle pravidla o umístění aminokyselin v globulárním řetězci se vyskytuje na povrchu proteinové globule, valin je hydrofobní aminokyselina, avšak v 3D uspořádání globinového řetězce u srpkovitosti se tímto způsobem objeví na povrchu. Zároveň je významné, že záporně nabitá kyselina glutamová je nahrazena alifatickou aminokyselinou bez náboje. Deoxygenovaná forma hemoglobinu (jak HbA, tak HbS) má poněkud jiné 3D uspořádání (viz výše), při němž se odkryje komplementární „lepivé“ místo, k němuž má valin v pozici 6 afinitu. Takto vzniklá molekula HbS vykazuje ve srovnání s HbA odlišné vlastnosti a ve své deoxygenované podobě se molekuly hemoglobinu shlukují a deformují krvinku (drepanocyt) (Obr. 48).
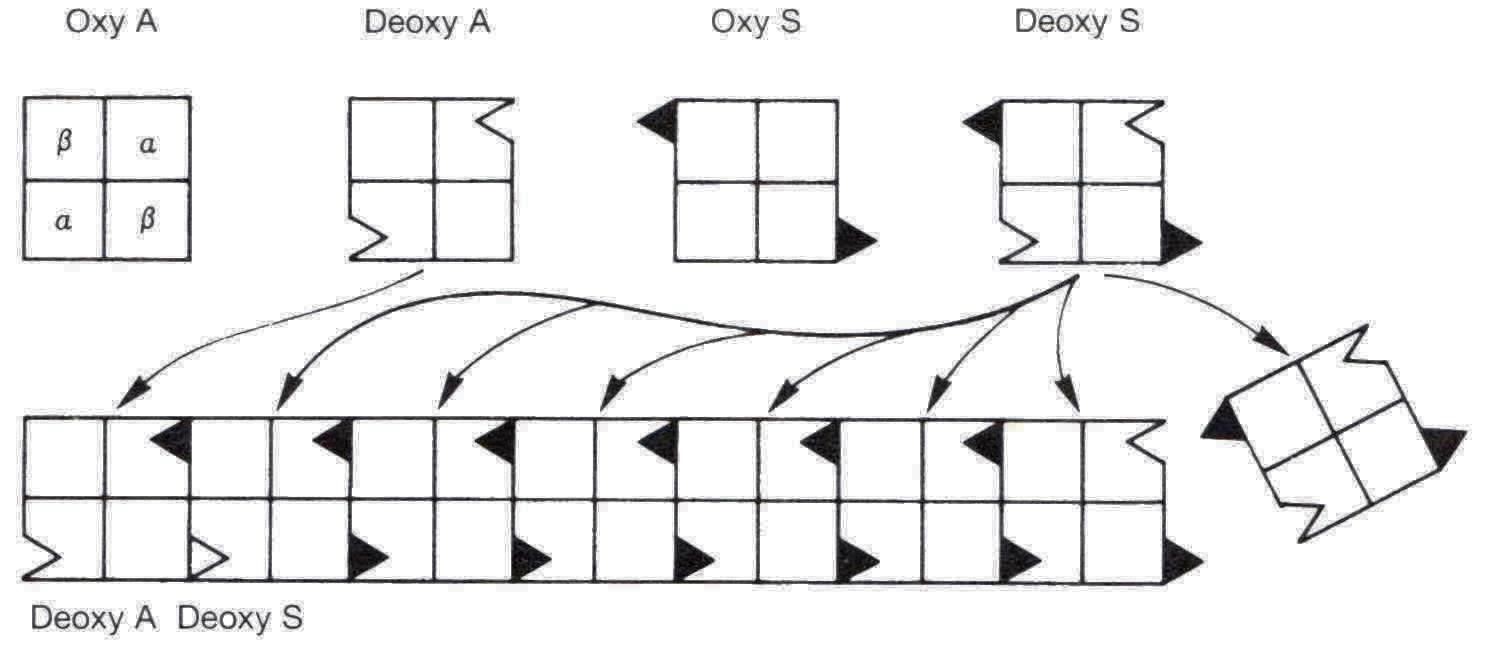
Obr. 48. Sřetězení molekul deoxyhemoglobinu u srpkovité anémie. (Murray et al. 1998)
Ačkoliv je drepanocyt menší než normální erytrocyt, není dostatečně pružný při průchodu malými cévami a vyvolává infarzaci.
Srpkovitá anémie se objevuje už v dětství, většinou u lidí (nebo jejich potomků) z tropických a subtropických pásem – tedy z regionů, ve kterých je běžná malárie (Obr. 49). Obyvatelé, kteří mají jeden gen ze dvou poznamenaný srpkovitou anémií, jsou proti malárii imunní (heterozygotní výhoda). Plasmodia totiž u malariků neprojdou erytrocytární fází. (Toto bylo po léta vysvětlováno změnou tvaru erytrocytu, ale mnohem významnější jsou pravděpodobně změněné vlastnosti plasmatické membrány drepanocytu.)
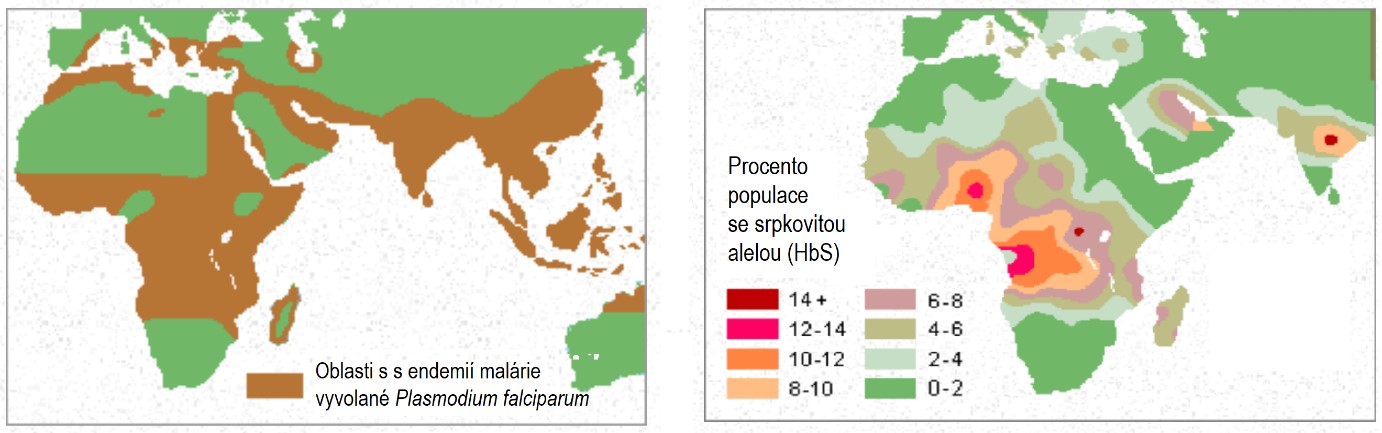
Obr. 49. Porovnání oblastí endemické malárie vyvolané Pl. Falciparum a výskytem alely pro HbS (upraveno z O‘Neil 1997)
Hemoglobinopatie C
Kromě srpkovité anémie existuje velké množství dalších hemoglobinopatií. Hemoglobinopatie C je zajímavá tím, že je postižen β-řetězec hemoglobinu ve stejném místě, jako u srpkovité anémie. Kyselina glutamová v poloze 6 od N-konce proteinu je však nahrazena lysinem, což znamená, že záporný náboj kyseliny glutamové je nahrazen dokonce kladným nábojem lysinu. HbC je méně rozpustný než HbA a krystalizuje v erytrocytech. Příznaky této hemolytické anémie jsou podobné, jako u srpkovité anémie.
Tyto typy hemoglobinopatií lze poměrně snadno laboratorně rozlišit elektroforeticky (Obr. 50 a 51).
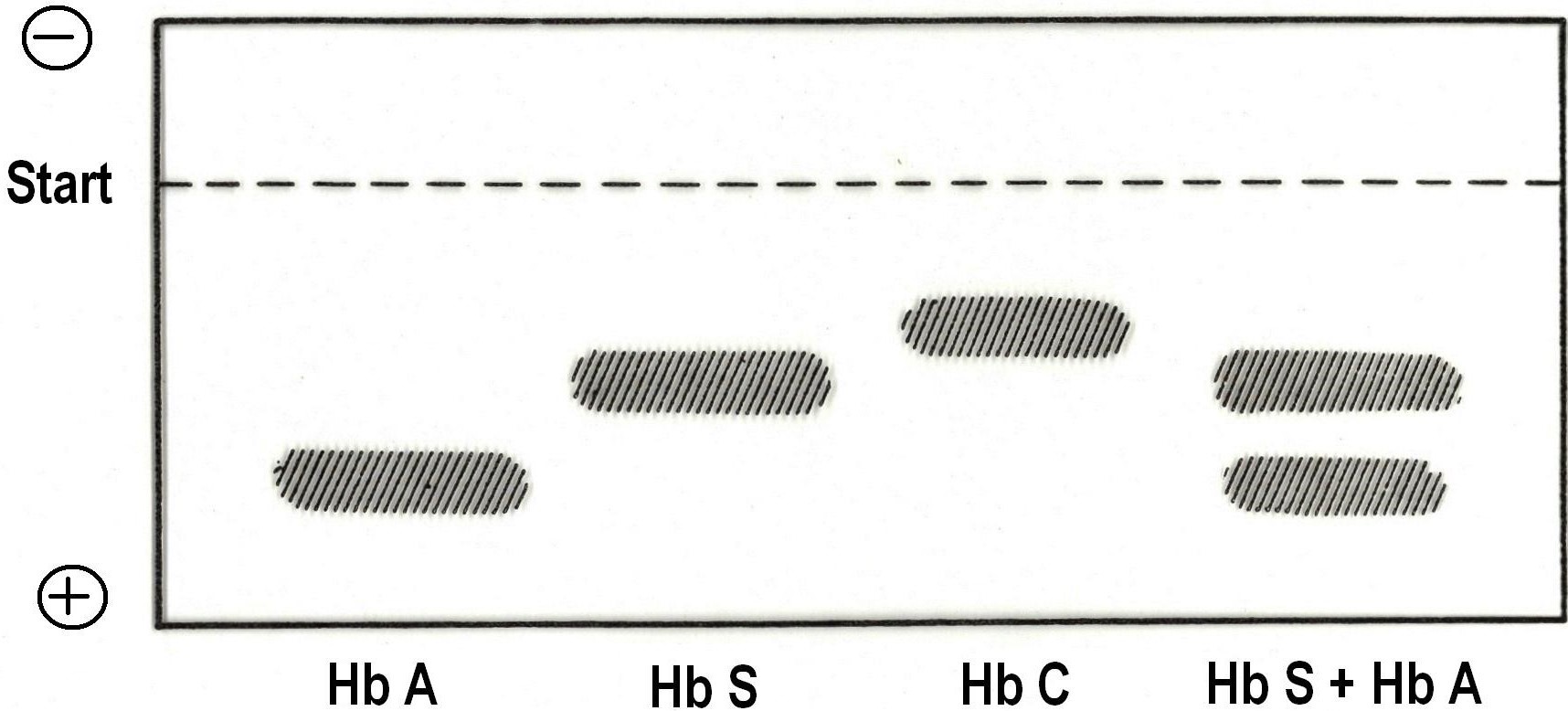
Obr. 50. Schéma elektroforetického rozlišení různých typů hemoglobinu (Dršata 1983)
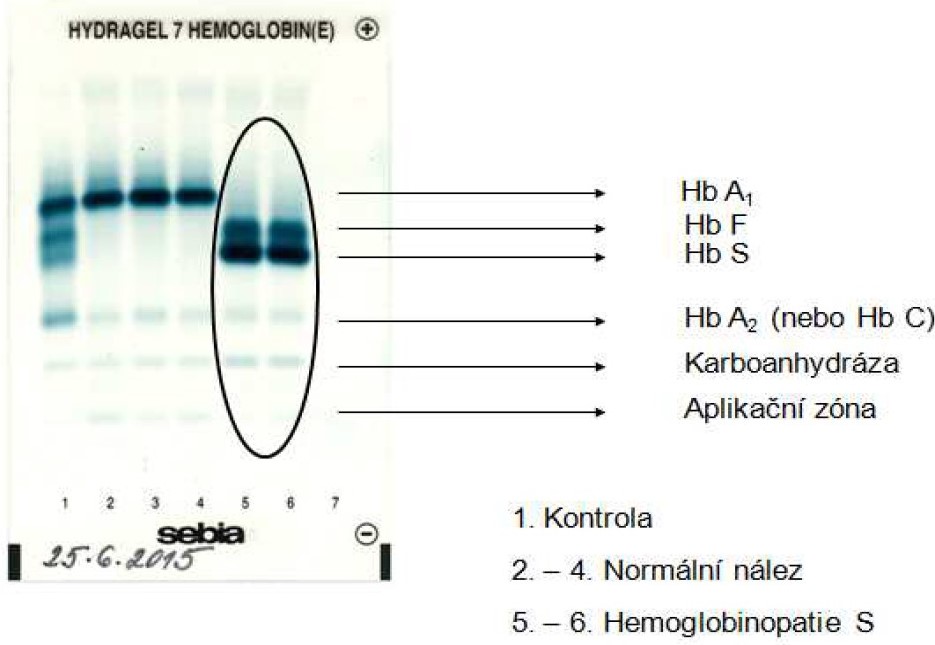
Obr. 51. Výsledek elektroforetické separace několika vzorků hemoglobinu (Rožcová 2016). Záznam pochází z laboratoře IV. Interní kliniky Fakultní nemocnice Hradec Králové.
Talasemie
Talasemie jsou nejčastější monogenně dědičná onemocnění. Jde o heterogenní skupinu chorob s poruchou syntézy jednoho z globinových řetězců. Druhý řetězec se syntetizuje v normálním množství. Protože je v relativním nadbytku, precipituje v erytrocytech, způsobuje jejich předčasnou destrukci a prohlubuje hypochromní anemii. Podle toho, jak podstatný je defekt syntézy jednoho z řetězců, může se jednat o klinicky bezvýznamný stav až po těžkou anemii.
Značný výskyt talasemií ve světě souvisí s tím, že poskytuje výhodu, protože heterozygoti talasemie jsou odolnější vůči malárii.
Normální hemoglobin se skládá ze stejného počtu globinových řetězců typu α a β, rozeznáváme poruchy syntézy α-globinového řetězce (α-talasemie) nebo β-globinového řetězce (β-talasemie).
Alfa-talasemie
K syntéze α-řetězců dochází na základě exprese 4 genů, které jsou umístěny na 16. chromosomu po dvou lokusech (viz obr. 39 na začátku této podkapitoly). Proto se α-talasemie vyskytuje ve čtyřech typech podle závažnosti defektu (Obr. 52).
- Delece jedné alely
Klinicky se neprojeví, jedná se o nosiče.
- Delece dvou alel
Stav se nazývá α-thalassemia minor a je bez podstatných příznaků, bývá lehká mikrocytární anemie.
- Delece tří alel
Jde o závažnou poruchu, nazývanou α-thalassemia intermedia nebo choroba hemoglobinu HbH. Tvoří se jen 25 % α-řetězců, čemuž odpovídá pouze malé množství HbA, HbF a HbA2. Výrazný relativní přebytek β-řetězců vede k tvorbě hemoglobinu H (HbH), složeného ze 4 řetězců β.
HbH má vysokou afinitu ke kyslíku. Řetězce se co do afinity ke kyslíku chovají samostatně, chybí Bohrův efekt kooperace řetězců (srovnej výše s obr. 45 ilustrujícím sycení fyziologických typů hemoglobinu kyslíkem). Afinita ke kyslíku je srovnatelná s afinitou myoglobinu a HbH proto nemůže působit jako účinný dodavatel kyslíku tkáním.
- Delece čtyř alel
Úplné chybění řetězců α. Tvoří se tzv. hemoglobin Bart (HbBart), složený ze čtyř řetězců γ (fetální řetězce). Tento stav je neslučitelný se životem, vzniká hydrops fetalis, dochází k úmrtí plodu.
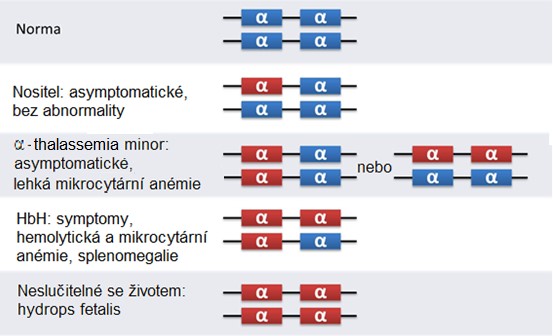
Obr. 52. Genetika α-talasemie a klinické následky. Červeně jsou označeny chybějící alely (upraveno z Razani 2010)
Beta-thalasemie
Postižena je syntéza β-řetězců, což se projeví až u hemoglobinu postnatálního. Na rozdíl od 4 alel pro řetězce α jsou u zdravého jedince k dispozici pouze 2 alely β. Zato na chromosomu 11 jsou alternativní geny pro řetězce γ a δ. Při poruchách syntézy β řetězců vzniká relativní nadbytek α, γ a δ řetězců.
Anemie se objeví až po 3 měsících života, kdy má být syntéza fetálního hemoglobinu vystřídána syntézou hemoglobinu HbA. Příčinou talasemie bývá mutace, kdy záměna báze v genetickém záznamu vede k vzniku stop-kodonu (Obr. 53).
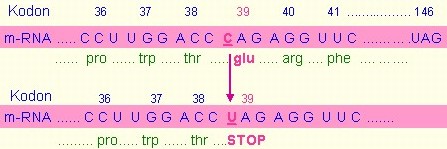
Obr. 53. Příklad bodové mutace v genu pro β-globinový řetězec, vedoucí k β-talasemii. Záměnou C v kodonu 39 (v tripletu CAG, kódujícím pro glutamát) za U vzniká stop-kodon UAG (upraveno z Cuschieri 2019)
Beta-thalasemie se vyskytuje ve třech typech podle závažnosti defektu (Tab. 7).
Tabulka 7. Přehled β-talasemií (upraveno z Chonat a Quinn 2017) - β0 – netvoří se žádný globin; β+ - mutovaný gen si zachoval částečnou funkci, tvoří se defektní β-řetězec; βE – bodová mutace, v pozici 26 je místo glutamátu lysin, typická mutace v populaci Indie a jihovýchodní Asie.

Beta-thalassemia minor
V tomto případě je postižena jen jedna alela genu pro syntézu řetězců β, druhá alela je normální. Obvykle se objevuje mírná asymptomatická hemolytická anemie, která se manifestuje během infekčního onemocnění, stresu nebo nedostatku kyseliny listové.
Beta-thalassemia intermedia
Může být způsobena heterozygotní mutací způsobující mírné snížení tvorby β-řetězců, případně homozygotní mutací s větším nedostatkem β-řetězců. Příznaky bývají různého stupně: anemie, žloutenka, splenomegalie, hepatomegalie. Příznaky se zhorší při stresu, infekci nebo nedostatku kyseliny listové (podobně jako u thalassemia minor).
Beta-thalassemia major
Jsou postiženy obě alely genu pro syntézu β-řetězců, produkce je podstatně omezena. To se projevuje jako těžká mikrocytová anemie. Nedostatek HbA je doplňován tvorbou HbF, který má příliš vysokou afinitu ke kyslíku (viz obr. 40 výše; tedy relativně méně ochotně uvolňuje kyslík pro tkáně) a není proto vhodný v postnatálním životě. Bývá opožděný vývoj, hepatosplenomegalie. mentální retardace. Nemocní umírají v mládí. Postižení musí být léčeni transfuzemi. Nejtěžší formou β-talasemie je Cooleyova (středomořská) anemie. Je to nejtěžší forma. U tohoto typu se vůbec nesyntetizují β-řetězce. Vyskytuje se proto HbA2 (α2δ2) a fetální hemoglobin HbF (α2γ2). Pro úplné chybění HbA se provádějí od narození krevní transfuze, avšak postižení umírají většinou v prvním roce života.