Metabolismus glykoproteinů a proteoglykanů a jeho poruchy
Glykoproteiny (mukoproteiny) a proteoglykany (mukopolysacharidy) patří mezi komplexní sacharidy. Základní rozdíly v jejich struktuře jsou shrnuty v Obr. 10.
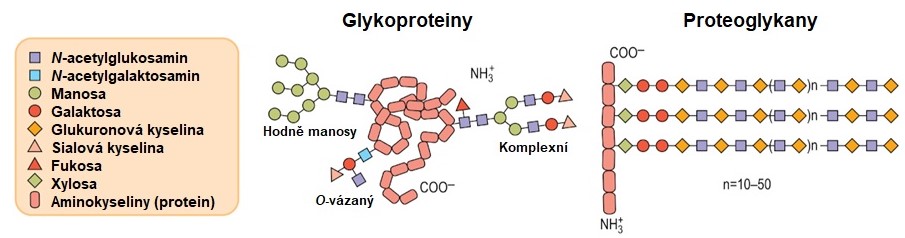
Obr. 10. Obecná struktura glykoproteinů a proteoglykanů (upraveno z Baynes a Dominiczak 2014)
Struktura a funkce glykoproteinů
Glykoproteiny jsou obvykle membránové proteiny nebo proteiny secernované z buňky obsahující cukernou složku, která je připojena jako krátká nebo dlouhá větev či nevětvený oligosacharidový řetězec. Tyto látky mají neutrální charakter a váhový podíl sacharidů nepřevyšuje podíl proteinu. Jedná se o nejčastější posttranslační modifikaci proteinů (zhruba 50 % eukaryotických proteinů má na sobě navázané oligosacharidy). Glykosylace, enzymové připojení sacharidových zbytků (neplést s glykací!), je katalyzováno glykosyltransferasami. Připojené cukerné řetězce (glykany) jsou kratší a obvykle jsou složené ze 2-10 cukerných zbytků. Sacharidy jsou na proteinový řetězec připojený kovalentní glykosidickou vazbou přes amidový dusík asparaginu (N-glykany) nebo prostřednictvím hydroxylové skupiny tyrosinu, threoninu nebo hydroxylysinu (O-glykany). Některé proteiny jsou v plasmatické membráně kotveny glykosylfosfatidylinositolovými strukturami (GPI-kotva). U těchto struktur je na vnější straně plasmatické membrány zanořený fosfatidylinositol s navázanými mastnými kyselinami a ten je spojen přes N-acetylglukosamin s glykanovým řetězcem, který je připojen amidovou vazbou fosfatidylethanolaminu na C-koncový aminokyselinový zbytek proteinu (Obr. 11). Počet glykanů připojených k jednomu řetězci proteinu kolísá od 1 do 30 i více. Glykany nejčastěji obsahují těchto osm cukerných zbytků: glukosu, galaktosu, manosu, N‑acetylglukosamin, N-acetylgalaktosamin, xylosu, fukosu a N‑acetylneuraminovou kyselinu (obvykle na koncích oligosacharidových řetězců). Pořadí cukerných zbytků je obdobou sekvence nukleotidů v DNA/RNA či aminokyselin v proteinech.
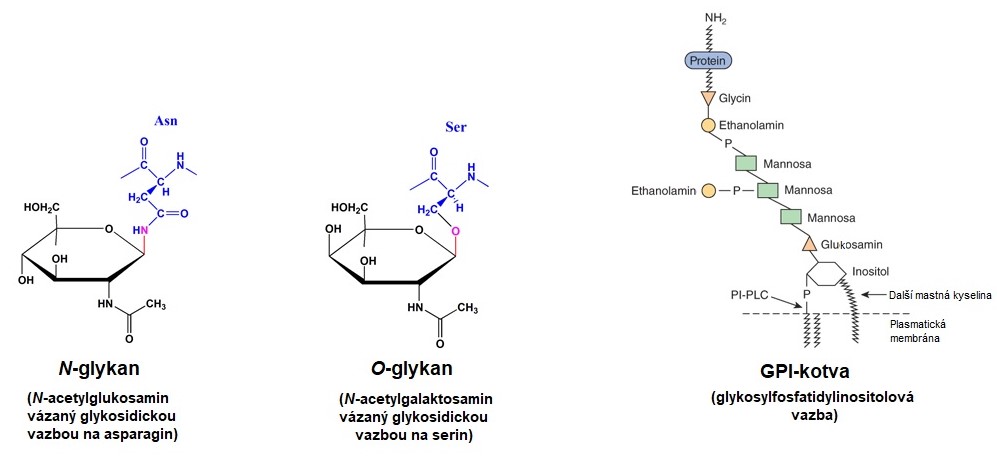
Obr. 11. Různé typy připojení glykanových řetězců na proteiny (upraveno z Wikimedia Commons 3, Wikimedia Commons 4 a Murray et al. 2012)
Glykoproteiny plní v organismu celou řadu funkcí (Obr. 12). Ovlivňují fyzikálně-chemické vlastnosti proteinů, jako jsou rozpustnost, náboj, viskozita, konformace, denaturace, vazebná místa pro různé molekuly, bakterie, viry a některé parazity. Chrání proteinovou část před proteolýzou uvnitř i vně buňky. Ovlivňují proteolytický sestřih prekurzorových proteinů na menší produkty. Uplatňují se v biologických aktivitách jako hormony (např. lidský choriogonadotropin) či enzymy (např. hydolasy, proteasy, glykosidasy, faktory hemokoagulace). Ovlivňují inserci do membrán, intracelulární transport, třídění a sekreci. Ovlivňují embryonální vývoj diferenciací. Umožňují interakci s lektiny. Slouží jako strukturní molekuly (např. kolagen, elastin, fibriny, kostní matrix). Pořadí cukerných zbytků v glykanech některých glykoproteinů na povrchu erytrocytů rozhoduje o krevní skupině.
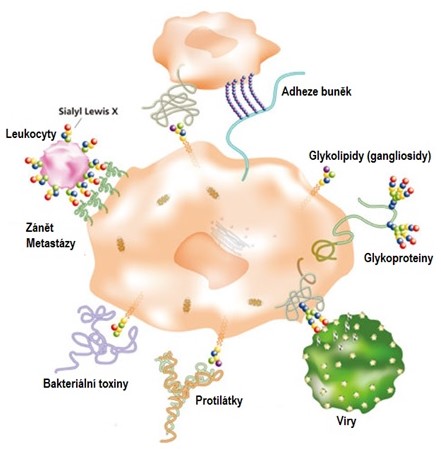
Obr. 12. Různé biologické procesy zprostředkované glykany na povrchu buněk (upraveno z Tsuji 2011).
Mezi glykoproteiny s O-vázanými oligosacharidovými řetězci patří mimo jiné muciny. Sekreční muciny se nacházejí v hlenu přítomném v sekretech GIT, respiračního a reprodukčního traktu (94 % vody, 5 % mucinů, zbytek tvoří směs elektrolytů, zbytků buněk a dalších látek). Tento hlen má vysokou viskozitu a často tvoří gel. Funkcí mucinů je zvlhčovat a tvořit ochrannou fyzikální bariéru na povrchu epitelu. Membránově vázané muciny se účastní různých mezibuněčných interakcí (např. selektiny - zánět). Muciny často maskují určité povrchové antigeny, např. nádorové buňky tvoří nadměrné množství mucinů, které je mohou ochránit před imunitní kontrolou. Muciny nesou specifické nádorové antigenní determinanty (peptidové a sacharidové epitopy rozpoznávané protilátkou), čehož bylo využito ke stimulaci imunitní odpovědi proti nádorovým buňkám.
N-vázané glykoproteiny jsou často studované, protože tato třída zahrnuje snadno dostupné glykoproteiny (např. plasmatické). Do této třídy patří nejen cirkulující glykoproteiny, ale i glykoproteiny membránově vázané. N-glykanové řetězce mají některé specifické funkce, např. mannosa-6-fosfátové zbytky jsou důležité pro směrování nově nasyntetizovaných glykoproteinů do organel (v tomto případě směrování lyzosomálních enzymů z Golgiho komplexu do lyzosomů). Velké N-glykanové řetězce se podílejí na udržení nově nasyntetizovaných glykoproteinů v rozpustném stavu v lumen endoplasmatického retikula. Jeden typ N-glykanových řetězců se účastní sbalování a zadržování některých glykoproteinů v lumen endoplasmatického retikula. Na tento typ N-glykanů se váže proteiny endoplasmatického retikula kalnexin a kalretikulin, které slouží jako chaperony a lektiny, a tak brání glykoproteinu v agregaci. Na komplex kalnexin-glykoprotein se váže proteindisulfidisomerasa ERp57, která katalyzuje tvorbu disulfidových můstků a tak usnadňuje správné sbalení glykoproteinu. Endoplasmatické retikulum pak opouští správně sbalený glykoprotein. Kalnexin a kalretikulin jsou důležitou součástí systému kontroly kvality fungující v lumen endoplasmatického retikula.
Třetí třídou glykoproteinů jsou GPI-struktury, které umožňují zakotvení proteinů do vnější vrstvy plasmatické membrány. Takovýmto způsobem jsou do plasmatické membrány ukotveny např. acetylcholinesterasa (membrány erytrocytů), alkalická fosfatasa (střevní, placentální) či prionový protein (neurony a další typy buněk). Ve srovnání s transmembránovými proteiny umožňuje GPI-kotva větší pohyblivost proteinů v plasmatické membráně. Některé GPI-kotvy mohou být spojené s intracelulárními signálními drahami. U některých polarizovaných epitelových buněk mohou GPI-kotvy odesílat některé proteiny do apikálních a basolaterálních domén plasmatické membrány.
Syntéza glykoproteinů je komplexní proces, který probíhá v endoplasmatickém retikulu a Golgiho komplexu a vyžaduje přítomnost specifických glykosyltransferas. Celý proces probíhá v několika fázích. Nejprve jsou v cytoplasmě vytvořeny aktivované sacharidy (např. GDP-mannosa, UDP-glukosa a UDP-N-acetylglukosamin), které jsou následně připojeny na dolicholfosfát a vznikající oligosacharidová struktura je odeslána do lumen endoplasmatického retikula. Zde dochází k postupnému sestavení („assembling“) glykanu se 14 cukernými zbytky přidáním dalších sacharidových zbytků. Následuje přemístění/připojení („processing“) tohoto prekurzoru k nascentnímu proteinu a transport do Golgiho komplexu, kde dojde ke konečnému zpracování s odstraněním některých sacharidových jednotek a připojením jiných.
Vrozené poruchy glykosylace
Abnormality v syntéze glykoproteinů a glykolipidů se uplatňují v rozvoji celé řady chorobných stavů označovaných souhrnně jako vrozené poruchy glykosylace (CDG). Jedná se o skupinu vzácných onemocnění, která jsou děděna autosomálně recesivně. Dosud bylo identifikováno 75 poruch a rozhodně se nejedná o konečné číslo. Ač jsou jednotlivě vzácná, jejich souhrnná incidence (1 : 20.000) je řadí mezi častá onemocnění. Jedná se o poruchy N‑glykosylace, O-glykosylace, O-mannosylace, kombinované poruchy N- a O-glykosylace a poruchy glykosylace lipidů. Rozlišujeme CDG typu I (poruchy sestavování, „assembling“) a CDG typu II (poruchy transportu, „processing“).
CDG mají velmi široké spektrum klinických projevů, zasahují totiž mnoho orgánů a symptomy se u jednotlivých onemocnění liší. Často se projevují jako různé neurologické poruchy, objevuje se u nich dysmorfie obličeje, zpomalený růst, sbíhavé šilhání, poruchy srážení krve, onemocnění jater a GIT.
K laboratorní diagnostice se nejčastěji používá isoelektrická fokusace s následnou imunofixací, která umožňuje detekci poruch N-glykanů (transferin) a umožňuje rozlišit CDG typu I a II (Obr. 13). Pro detekci poruch O-glykanů slouží analýza sérového apoCIII pomocí isoelektrické fokusace s následným western blottingem. Pro diagnostiku CDG syndromu lze použít i turbidimetrické stanovení relativního poměru mezi celkovým množstvím transferinu a asialovanými, monosialovanými a disialovanými transferiny. Pro přesné určení typu onemocnění je nutná enzymová analýza (pokud je pro daný typ CDG dostupná), strukturní analýza oligosacharidů vázaných na lipid a/nebo N-vázaných glykoproteinů v kultivovaných fibroblastech a molekulárně-genetické vyšetření.
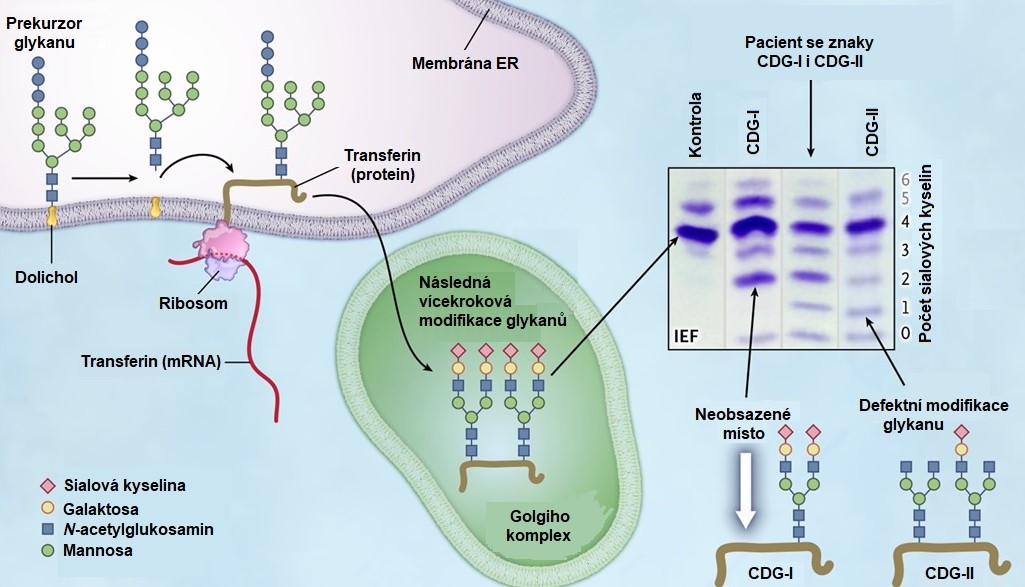
Obr. 13. Biosyntéza glykoproteinu transferinu a laboratorní diagnostika vrozených poruch N‑glykosylace (upraveno z Tegtmeyer et al. 2014).
Struktura a funkce proteoglykanů
Proteoglykany jsou struktury s proteinovým jádrem, na které jsou kovalentně vázané sacharidové jednotky zvané glykosaminoglykany (GAG). Dosud bylo popsáno minimálně 30 proteoglykanů (např. agrekan, fibromodulin, dekorin), které se liší tkáňovou distribucí, funkcí a složením (tj. povahou proteinového jádra a připojenými GAGy). Tyto struktury se tvoří významnou část mezibuněčné hmoty (ECM) a zastávají celou řadu biologických funkcí.
Součástí proteoglykanů jsou GAGy, které se však mohou vyskytovat i volně. Zatím bylo identifikováno sedm GAGů: kyselina hyaluronová, chondroitinsulfát, dermatansulfát, keratansulfát I a II, heparin a heparansulfát (Obr. 14). Jedná se o nevětvené polysacharidy složené z opakujících se disacharidových jednotek, které tvoří aminocukr (D-glukosamin, D‑galaktosamin) a uronová kyselina (kys. idurunová, galakturonová či glukuronová) s výjimkou keratansulfátu, kde je místo uronové kyseliny přítomná galaktosa. Sulfátová skupina je připojena na všechny GAGy s výjimkou kyseliny hyaluronové a to buď ve formě O‑esteru, nebo N-sulfátu.
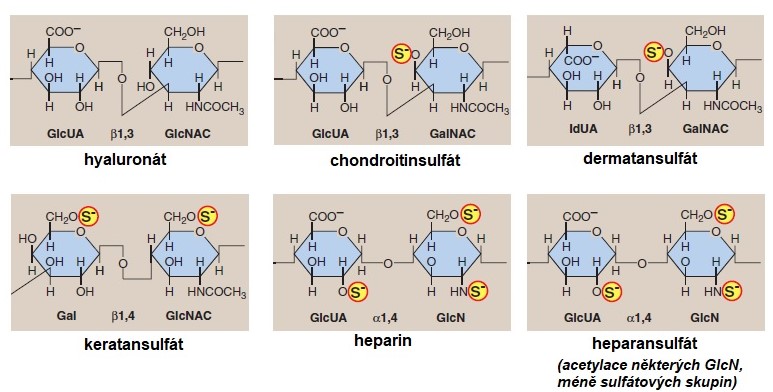
Obr. 14. Struktura známých glykosaminoglykanů (upraveno z Harvey a Ferrier 2011)
GAGy se vzájemně liší svou strukturou, tkáňovou distribucí a biologickou funkcí. Kyselina hyaluronová se vyskytuje v bakteriích a v ECM prakticky všech živočišných tkání. Hojná je zejména v hydratovaných tkáních, jako je kůže a pupeční šňůra, dále v kostech, chrupavkách, synoviální tekutině kloubů a ve sklivci oka. Díky její schopnosti přitahovat vodu dochází k rozvolnění ECM, což umožní buňkám během morfogeneze a při hojení poranění migrovat. Vysoká koncentrace kyseliny hyaluronové a chondroitinsulfátu přispívá k elasticitě a odolnosti chrupavek vůči tlaku. Chondroitinsulfáty (sulfoskupina v pozici 4 nebo 6) jsou hlavní složkou chrupavek a jsou zodpovědné za udržení struktury ECM. Nacházejí se v kosti v místech kalcifikace, kde pravděpodobně váží vápenaté soli. Ve vysokém množství jsou přítomné rovněž v mezibuněčné hmotě CNS, kde kromě strukturní úlohy působí jako signální molekuly v prevenci opravy nervových zakončení po poranění. Keratansulfát I a II se liší ve způsobu připojení na osový protein a jejich distribuce není specifická pro určité tkáně. V oku se nacházejí mezi fibrilami kolagenu a jsou nezbytné pro udržení průhlednosti rohovky. Dermatansulfát je přítomen prakticky ve všech tkáních a je hlavním GAGem v kůži. Pravděpodobně se podílí na srážení krve, hojení ran a odolnosti vůči infekci. Heparin se nachází v granulech mastocytů a také v játrech, plicích a kůži. Interaguje s plasmatickým antithrombinem a faktory IX a XI krevního srážení, takže působí antikoagulačně. Heparin se specificky váže na lipoproteinovou lipasu ve stěně kapilár a způsobuje uvolnění tohoto enzymu do cirkulace. Heparansulfát je přítomný na extracelulárním povrchu celé řady buněk a je asociován s jejich plasmatickou membránou. Působí pravděpodobně jako receptory a podílí se zřejmě na buněčném růstu a mezibuněčné komunikaci. Spolu s kolagenem typu IV a lamininem je přítomen rovněž v bazální membráně ledvin, kde určuje nábojovou selektivitu při glomerulární filtraci. Některé GAGy se používají v terapii jako léčiva – k léčbě osteoartrózy se používají kyselina hyaluronová a chondroitinsulfát (dále též glukosaminsulfát a glukosaminhydrochlorid); heparin a heparansulfát (heparinoidy – např. danaparoid) působí jako antitrombotika a snižují krevní srážení.
Proteoglykany jsou komplexní molekuly se složitou strukturou a obsahují větší množství sacharidů než glykoproteiny a to až 95 % své hmotnosti. Proteiny, které jsou kovalentně připojené ke GAGům, se nazývají osové proteiny. Struktura proteoglykanu agrekanu, který se vyskytuje v chrupavkách kostí, je na Obr. 15. Jeho molekula připomíná kartáč na lahve – obsahuje dlouhé řetězce kyseliny hyaluronové s nekovalentně připojenými spojovacími proteiny. Ty nekovalentně interagují s osovými proteiny, na které jsou napojené řetězce GAGů keratansulfátu a chondroitinsulfátu. Díky velkému počtu připojených sulfátových skupin mají proteoglykany často záporný náboj.
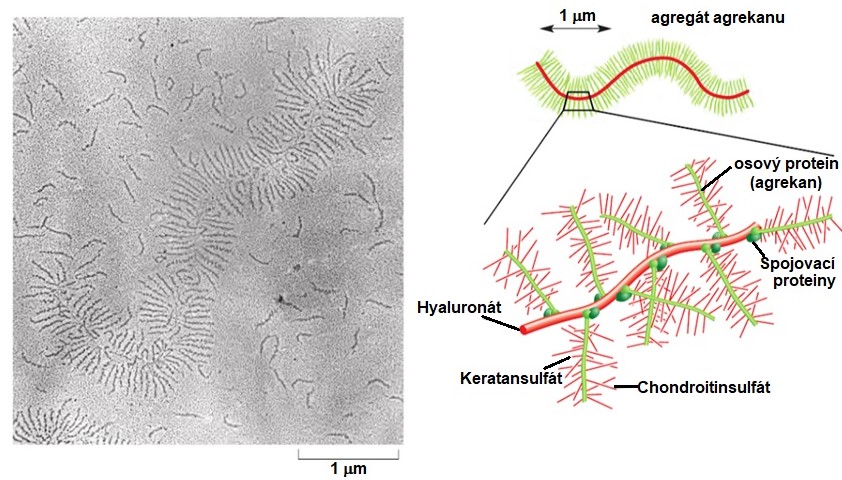
Obr. 15. Struktura proteoglykanu agrekanu (upraveno z Alberts et al. 2007)
Proteoglykany se vyskytují ve všech tkáních těla a to zejména v ECM, kde asociují navzájem a specifickým způsobem také s dalšími strukturními složkami (např. kolagenem a elastinem). Interakce proteoglykanů s kolagenem a elastinem jsou důležité pro určení strukturní organizace matrix ECM. Některé proteoglykany interagují s adhezivními proteiny ECM (laminin, fibronektin, vitronektin). Jiné jsou schopné vázat růstové faktory (např. TGF-β) a tím ovlivňovat jejich účinek na buňky. Díky záporným nábojům GAGů jsou proteoglykany polyanionty, které váží polykationty (např. Na+, K+) a tak přitahují do ECM osmotickým tlakem vodu a přispívají k jejímu turgoru. Díky struktuře GAGů a jejich schopnosti vytvářet již při nízkých koncentracích gel působí proteoglykany jako filtr, který umožňuje relativně volnou difuzi malých molekul a omezuje vstup velkých makromolekul do ECM. Ve srovnání s proteiny zabírají proteoglykany v matrix ECM díky své struktuře a schopnosti tvořit agregáty větší objem.
Mukopolysacharidosy
Mukopolysacharidosy (MPS) jsou heterogenní skupinou onemocnění vyvolaných deficitem některého enzymu odbourávajícího GAGy. Degradace GAGů probíhá v lyzozomech postupným působením celé řady hydrolas, která zahrnují endoglykosidasy, exoglykosidasy a sulfatasy. Při deficitu některého enzymu dochází ke střádání nerozložených a neodstraněných GAGů uvnitř lyzosomu buněk četných tkání (např. játra, slezina, kosti, kůže, CNS). Tato onemocnění, která patří mezi lyzosomální střádavé poruchy, jsou děděna nejčastěji autosomálně recesivně s výjimkou Hunterovovi nemoci, jejíž dědičnost je vázána na chromosom X.
Do současné doby bylo identifikováno sedm MPS, z nichž některé mají několik podtypů. Klinické projevy jsou značně různorodé a závisí na typu konkrétního defektu (Tab. 2). Souhrn hlavních znaků MPS zahrnuje chronický progresivní průběh onemocnění, multiorgánové poškození, častá organomegalie (např. splenomegalie, hepatomegalie), závažné abnormality skeletu, faciální dysmorfie, dále abnormality sluchu, zraku, kardiovaskulárního systému a mentálního vývoje. Může dojít ke zkrácení předpokládané délky života či k regresi psychomotorického vývoje.
Podle zbytkové aktivity defektní α-L-iduronidasy se rozlišují tři typy MPS I, které se označují jako syndrom Hurlerové, syndrom Hurlerové-Scheie a syndrom Scheie. Nejtěžší formou je syndrom Hurlerové, kde je zbytková aktivita tohoto enzymu prakticky nulová. V důsledku enzymového defektu se hromadí dermatan- a heparansulfát. Toto onemocnění se projevuje malým vzrůstem postižených, hepatosplenomegalií a závažnou kraniofaciální dysmorfií. Díky hrubým rysům v obličeji bylo toto onemocnění označováno jako gargoylismus (gargoyl = chrlič). U pacientů se často opakují ORL infekce a infekce dýchacích cest. Kompletní klinický obraz onemocnění se zpravidla rozvíjí ve 2.-3. roce života. Objevují se kostní deformity až dysostosis multiplex, progresivní poruchy učení, zpomalení psychomotorického vývoje a progresivní ztráta osvojených dovedností. Častá je hluchota následkem opakovaných infekcí uší a zákal rohovky. Ve školním věku se přidává kardiomyopatie a postižení chlopní. Neléčení pacienti umírají před dosažením 10 let věku na kardiopulmonální onemocnění. Syndrom Hurlerové-Scheie a syndrom Scheie jsou lehčí formy MPS I s částečným deficitem enzymu, u nichž bývá postižení skeletu i intelektu menšího rozsahu. K léčbě MPS I se používá transplantace hematopoetických kmenových buněk a enzymová substituční terapie.
Hunterův syndrom je jedinou MPS s gonosomálně recesivní dědičností (postižení pouze chlapci). Toto onemocnění je vyvoláno deficitem iduronát-2-sulfatasy s následným hromaděním dermatan- a heparansulfátu, které se v nadbytku vylučují močí. MPS II má mnoho společných symptomů s MPSI. Typická je přítomnost modrých mongolských skvrn na kůži, pupeční či tříselná kýla, změny vzhledu obličeje, vystouplé břicho opakující se infekce dýchacích cest a uší (může vést ke hluchotě), chronická rýma a psychomotorická retardace. U těžších forem onemocnění se objevují poruchy chování s hyperaktivitou, které se stoupajícím věkem mizí (ztráta fyzických i psychických sil). Neléčení pacienti umírají obvykle kolem 15 let věku na srdeční selhání. Mezi používané léčebné přístupy patří enzymová substituční terapie a transplantace hematopoetických kmenových buněk či kostní dřeně.
Syndrom Sanfilippo je poruchou katabolismu heparansulfátu s primárním postižením CNS. Podle defektního enzymu dělíme tento syndrom na čtyři podtypy (MPS IIIA sulfamidasa; IIIB α-N-acetylglukosaminidasa; IIIC α-glukosaminid-N-acetyltransferasa; IIID N-acetylglukosamin-6-sulfatasa). Jedná se o velmi závažná onemocnění, která jsou dosud neléčitelná. Onemocnění má tři fáze. V první fázi (předškolní děti) dochází k opoždění vývoje, poruchám sluchu a řeči. Dítě je sociálně méně přizpůsobivé a vzhledem k normálnímu vzhledu je diagnosa stanovena většinou velmi pozdě. V druhé fázi (3-10 let věku) jsou děti extrémně hyperaktivní, neklidné, objevují se poruchy chování a poruchy spánku (spí 2-4 hodiny denně), dostavuje se inkontinence. V poslední fázi dochází k progresivní ztrátě osvojených dovedností s postupnou deteriorací do vegetativního stavu. Průměrný věk dožití je 14 let.
U syndromu Morquio existují dva podtypy podle zasaženého enzymu (IVA galaktosamin-6-sulfatasa; IVB β-galaktosidasa). U obou typů se hromadí keratansulfát, v případě MPS IVA navíc ještě chondroitin-6-sulfát. Onemocnění je charakterizováno závažnými kostními deformitami (kostní dysplazie, vychýlení obratlů s kompresí míchy a ochrnutím), malým vzrůstem (95-105 cm), velkou kazivostí zubů, postižením rohovky a možnou ztrátou sluchu. Inteligence je normální a CNS není postižen. Léčba je obtížná – transplantace hematopoetických buněk není doporučena a enzymová substituční terapie je ve fázi výzkumu.
Syndrom Maroteaux-Lamy (MPS VI) je způsoben deficitem N-acetylgalaktosamin-4-sulfatasy, v důsledku čehož se hromadí dermatansulfát. Somatické rysy nemocných nápadně připomínají MPS I ovšem bez výraznějšího postižení intelektu. Objevuje se kraniofaciální dysmorfie, deformity skeletu, hepatosplenomegalie, kardiomyopatie a postižení chlopní. Je dostupná enzymová substituční terapie.
Syndrom Sly (MPS VII) je vyvolán deficitem β-glukuronidasy, která normálně odštěpuje kyselinu glukuronovou od dermatansulfátu, heparansulfátu i chondroitinsulfátu (4- i 6-sulfátu). U nemocných se všechny čtyři GAGy hromadí a jsou ve zvýšené míře exkretovány močí. Z klinického hlediska se jedná o značně variabilní poruchu, která se nejčastěji projevuje jako hydrops fetalis. U pacientů, kteří přežijí těhotenství, je klinický obraz podobný MPS I, ale intelekt nebývá zasažen.
Nejvzácnější MPS je syndrom Natowicz (MPS IX), který byl poprvé popsán až v roce 1996. Onemocnění je způsobeno deficitem hyaluronidasy I a dochází u něj k hromadění kyseliny hyaluronové v tkáních. Mezi klinické projevy patří bolesti a otoky kloubů, změny pojivové tkáně (zmnožení) a mírná kraniofaciální dysmorfie. Intelekt je normální a zrakové funkce nejsou zasaženy.
Tab. 2. Přehled mukopolysacharidos a jejich hlavních klinických příznaků
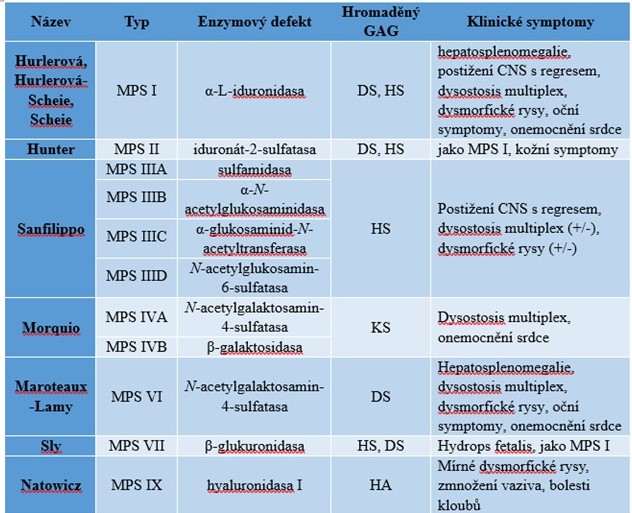
MPS, mukopolysacharidosa; GAG, glykosaminoglykan; DS, dermatansulfát; HS, heparansulfát; KS, keratansulfát; HA, kyselina hyaluronová; oční symptomy – zákaly rohovky a oftalmoplegie; dysostosis multiplex – kostní dysplazie se zhrubnutím obličejových rysů typická pro MPS
K laboratorní diagnostice těchto onemocnění se nejčastěji používá analýza moči. U kvantitativní analýzy moči se kolorimetrickou metodou (nejčastěji s dimethylmethylenovou modří) stanovuje množství vylučovaných GAGů s ohledem na věk pacienta, protože množství exkretovaných GAGů se s věkem snižuje (dospělí jedinci vylučují méně než novorozenci). Kvalitativní analýza se používá k určení jednotlivých exkretovaných GAGů a provádí se buď pomocí tenkovrstvé chromatografie, nebo elektroforézy (Obr. 16). Elektroforézou je možné analyzovat GAGy i v tkáňové biopsii. V leukocytech a fibroblastech je stanovována katalytická aktivita příslušného enzymu. K potvrzení diagnosy se využívá analýza DNA. U většiny MPS je dostupná prenatální diagnostika s použitím buněk plodové vody či biopsie choriových klků.
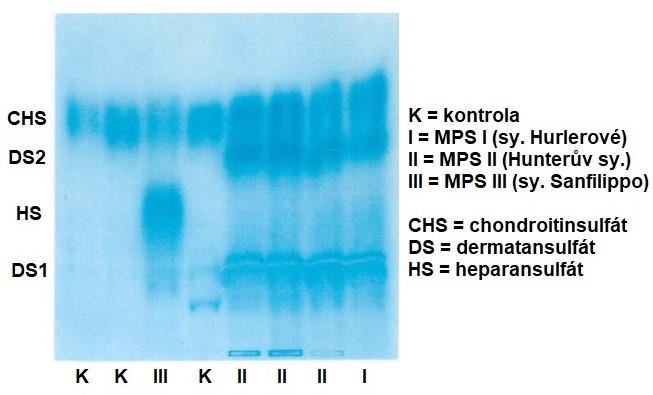
Obr. 16. Analýza exkrece dermatansulfátu a heparansulfátu v moči (převzato z Ledvinová 2015).
Léčba MPS je obtížná a často je dostupná pouze symptomatická léčba, která umožňuje zmírnit následky onemocnění. Pacienti jsou v péči celé řady specialistů (např. ortopedů, stomatologů, otorhinolaryngologů, kardiologů, očních lékařů, pneumologů, neurologů), kteří zajišťují ortopedickou korekci kostních změn, dechovou rehabilitaci, oční a sluchové korekce či medikamentózní léčbu nespavosti či hyperaktivity. Celková léčba je velmi nákladná (často se pohybuje v milionech Kč/rok) a jednotlivé léčebné přístupy se volí podle typu onemocnění, věku pacienta, závažnosti a typu klinických obtíží. Mezi další terapeutické přístupy používané v léčbě MPS patří transplantace hematopoetických buněk, substrát-redukční terapie (SRT) a enzymová substituční terapie (ERT). Transplantace hematopetických kmenových buněk se využívá k léčbě MPS I a MPS VI, přináší však s sebou riziko vzniku reakce štěpu proti hostiteli, která ohrožuje život pacienta. Při enzymové substituční terapii je defektní enzym nahrazen lidským rekombinantním enzymem, který je podáván infuzí v pravidelných intervalech (obvykle 1x týdně) a to po celý život. ERT je zatím dostupná k léčbě MPS I, II a VI, ale zkouší se i u MPS IV a VII. Nevýhodou tohoto typu léčby je, že rekombinantní enzym neprochází hematoencefalickou bariérou, a díky tvorbě protilátek proti podávanému enzymu může dojít k anafylaktické reakci. Substrát‑redukční terapie je založená na působení malých molekul, jež přímo inhibují klíčový enzym zapojený do biosyntézy hromadícího se substrátu. Příkladem může být použití flavonoidu genisteinu u MPS III, kde tato látka inhibuje proteinkinasovou aktivitu receptoru pro epidermální růstový faktor, která je nutná pro plnou expresi genů kódujících enzymy syntézy GAGů. U tohoto typu MPS, který postihuje především CNS, nepřináší transplantace hematopoetických kmenových buněk ani ERT kýžený efekt.