Poruchy metabolismu lipoproteinů
Dyslipoproteinemie a dyslipidemie
Název těchto poruch je odvozen od hodnot lipoproteinů nebo lipidů v krvi. Dříve se nejvíce používal název hyperlipoproteinemie, protože zvýšení koncentrace lipoproteinů v krvi je mnohem častější než jejich snížení (hypolipoproteinemie). Často je však zvýšení jedné frakce lipoproteinů provázeno snížením jiné nebo modifikací částic, proto se dnes spíše používá název dyslipoproteinemie nebo dyslipidemie.
Tak jako u řady jiných poruch a chorob lze použít i u dyslipoproteinemií klasifikaci podle příčiny. Podle toho rozlišujeme primární a sekundární poruchy.
Primární dyslipoproteinemie jsou geneticky podmíněné. Rozlišuje se např. familiární hypercholesterolémie nebo familiární hypertriacylglycerolémie.
Sekundární dyslipoproteinemie jsou důsledkem jiného onemocnění, které narušuje lipidový a lipoproteinový metabolismus. Mohou se projevit izolovaným zvýšením cholesterolu nebo triacylglycerolů nebo obou parametrů. Často doprovází např. diabetes mellitus, hypotyreózu, choroby jater, obezitu, chronický alkoholismus. Jejich nebezpečí spočívá v dlouhodobém bezpříznakovém období, k náhlé manifestaci pak dochází ve formě komplikace aterosklerózy nebo jako akutní hemoragická pankreatitida.
Terapeutická klasifikace
Terapeutická (klinická) klasifikace dyslipidemií je zavedena podle Evropské společnosti pro aterosklerózu. Představuje jednoduché a praktické rozdělení dyslipidemií na tři skupiny na základě stanovení koncentrace pouze základních lipidových složek, tj. cholesterolu a triacylglycerolů v séru nebo plazmě. Tato klasifikace je pak základem pro rozhodování o terapeutickém postupu.
Terapeutická klasifikace rozlišuje v případě hyperlipidemií tři možnosti:
- izolovaná hypercholesterolemie
- izolovaná hypertriacylglycerolemie
- kombinovaná (smíšená) hyperlipidemie
Výhodou této klasifikace je její snadná aplikace, snadné rozhodování o terapii. Tato klasifikace ovšem podstatně zjednodušuje problematiku. Nezohledňuje význam frakcí, rozlišuje rozdělení cholesterolu do jednotlivých frakcí s odlišným významem (LDL-HDL).
Z tohoto hlediska je na úrovni českého zdravotnictví již překonaná. Sice se v literatuře uvádí, že tato klasifikace je používána pro terapeutický postup, avšak při dnešním vybavení a standardních postupech klinickobiochemických laboratoří je už v počátečním vyšetření lipidového metabolismu rozlišován celkový cholesterol a LDL-cholesterol, což nejen podstatně pomáhá ve specifikaci terapeutického postupu, ale je i v povědomí české laické veřejnosti.
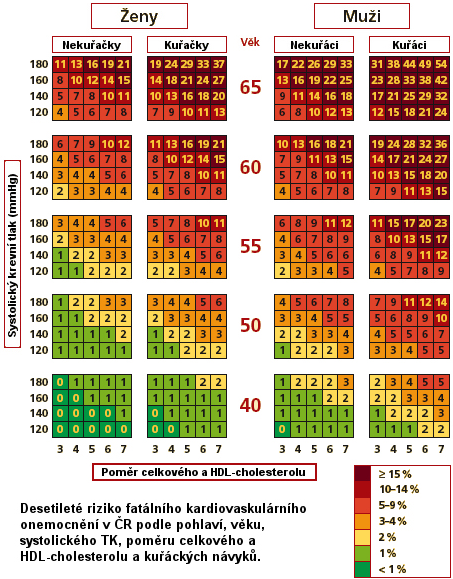
Tabulky České společnosti pro aterosklerózu, znázorňující riziko následků aterosklerózy ve vztahu základních rizikových faktorů aterosklerózy včetně frakcí cholesterolu jsou běžně vyvěšeny v ordinacích našich praktických lékařů (Tab. 5).
Tabulka 5. Desetileté riziko fatálního kardiovaskulárního onemocnění zahrnující základní faktory (podle Cífková et al. 2005)
Izolovaná hypercholesterolemie
Charakteristické pro tento typ je izolované zvýšení celkového cholesterolu, především v LDL frakci. Obvykle se rozlišuje familiární hypercholesterolemie a polygenní hypercholesterolemie.
- Familiární hypercholesterolemie je autosomálně dominantní onemocnění, jehož příčinou je genetická porucha v tvorbě nebo funkci LDL-receptorů. U homozygotů je katabolismus LDL pomocí LDL-receptorů prakticky nefunkční, u heterozygotů je kapacita LDL-receptorů snížena na polovinu. V důsledku toho se v krvi hromadí aterogenní LDL částice. Heterozygotní forma je častější a vyskytuje se asi 1 případ na 500 osob. Homozygoti jsou těžce postiženi již od dětství, vyskytují se u nich šlachové a kožní xantomy a většina z nich umírá na infarkt myokardu do 20 let. U postižených heterozygotních osob se manifestuje předčasným výskytem kardiovaskulárních onemocnění (ICHS ve věku 30–50 let). Změny v lipoproteinovém spektru odpovídají převážně fenotypu IIa, méně často IIb podle Fredricksona (viz dále).
- U polygenní hypercholesterolemie se uplatňují jak genetické vlivy, tak i vlivy prostředí. V průmyslově vyspělých zemích se s ní setkáváme velmi často. Hodnoty celkového cholesterolu nepřesahují obvykle 8 mmol/l, ale již představují zvýšené riziko aterosklerózy. Změny v lipoproteinovém spektru zde rovněž odpovídají převážně fenotypu IIa, méně často IIb podle Fredricksona.
Izolovaná hypercholesterolemie může být i sekundárním projevem např. u hypotyreózy, nefrotického syndromu nebo při stravě bohaté na nasycené tuky.
Kombinovaná hyperlipidemie
Tento typ představuje současné zvýšení cholesterolu i triacylglycerolů.
- Familiární kombinovaná hyperlipidemie patří k nejčastějším primárním hyperlipoproteinemiím. Vyskytuje se ve frekvenci 1:50 až 1:100. Má podklad v geneticky podmíněné zvýšené tvorbě apolipoproteinu B-100. Je spojena se zvýšeným rizikem vaskulárních onemocnění. Bývají zvýšeny LDL a VLDL, odpovídající fenotypu IIb podle Fredricksona, ale někdy i fenotypům IIa, IV a V.
- Sekundární kombinované formy bývají např. u hypotyreózy nebo při léčbě kortikoidy.
Izolovaná hypertriacylglycerolemie
U tohoto typu jde o izolované zvýšení triacylglycerolů:
- Geneticky podmíněnou hypertriacylglycerolémií je familiární hypertriacylglycerolemie, která postihuje asi 0,2–0,3 % populace. Projevuje se zmnožením VLDL, pravděpodobně na podkladě jejich zvýšené tvorby. Současně nacházíme sníženou hladinu HDL-cholesterolu. V laboratorním nálezu se setkáváme s mírně zvýšenými triacylglyceroly, obvykle do 6 mmol/l při normální koncentraci cholesterolu. U nemocných je nebezpečí infarktu myokardu.
- Vzácně se můžeme setkat s familiární hyperlipoproteinemií typu I (odpovídá typu I podle Fredricksona), charakterizovanou hyperchylomikronemií. Nemocní jsou ohroženi pankreatitidou vyvolanou vysokou hladinou triacylglycerolů, která zvyšuje její riziko.
- Sekundární forma hypertriacylglycerolémie je často spojena s diabetem mellitem, obezitou, nadměrným příjmem alkoholu nebo stravy s vysokým obsahem sacharidů.
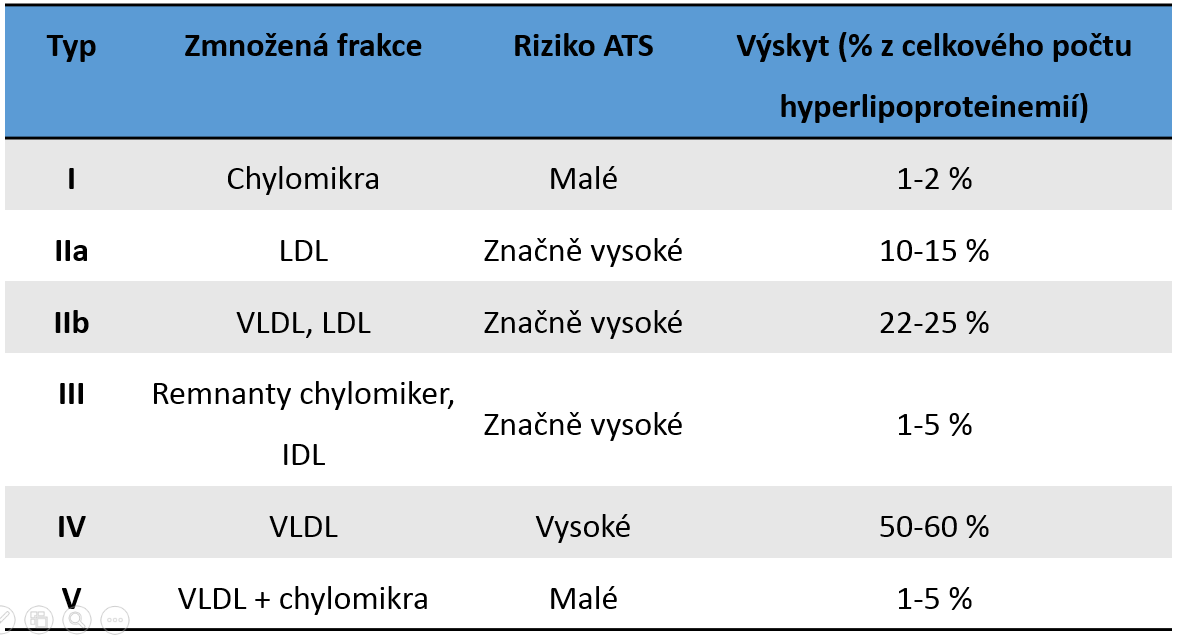
Jinou známou a dlouho používanou klasifikací je Fredicksonova (fenotypová) klasifikace, založená na elektroforetickém dělení lipoproteinů (Tab. 6). Vychází tedy stejně jako terapeutická klasifikace z laboratorního nálezu. V běžné praxi je považována za překonanou, avšak jak je zřejmé z předchozích i následujících odstavců, současná klasifikace primárních dyslipoproteinemií se na ni stále odvolává a liší se od ní pouze částečně.
Fredricksonova klasifikace není založena primárně na hledání příčin, avšak umožňuje některé typy dyslipoproteinemií popsat na molekulární úrovni.
Tabulka 6. Přehled Fredricksonovy fenotypové klasifikace hyperlipoproteinemií
Současná klasifikace primárních dyslipoproteinemií
A. Familiární hypercholesterolemie
Jako familiární hypercholesterolemie (též primární hypercholesterolemie) se označuje skupina tří poruch, které vedou k poruše vychytávání částic LDL prostřednictvím LDL‑receptoru (LDL-R). V důsledku toho se LDL částice hromadí v krevní plazmě, tj. stoupá LDL‑cholesterol. Porucha vede k závažné izolované hypercholesterolémii (s prakticky normálními hladinami HDL-cholesterolu a triacylglycerolů). Bez adekvátní léčby způsobuje akcelerovanou aterosklerózu, která často vede k vážné až fatální koronární ischémii v mladém věku (mnohdy do 3. decennia).
Příčinou může být:
- porucha LDL-receptoru,
- porucha apoproteinu B-100 (zpravidla záměna Arg3500 za Gln), který slouží jako ligand pro LDL-receptor,
- mutace genu pro proprotein konvertasu subtilisin/kexin 9 (PCSK9).
Dědičnost je ve všech třech případech autozomálně dominantní, přičemž hypercholesterolémie je samozřejmě těžší u homozygotů než u heterozygotů.
Celková incidence v evropské populaci a v USA se pohybuje kolem 1:200.
Ad 1) Poruchy LDL-receptoru (odpovídá typu Fredrickson IIa)
Jde o primární hypercholesterolemii, která se dědí autosomálně dominantně. Incidence heterozygotů je 3–4 na 1000 obyvatel a homozygotů 3–4 na 1 milion. Příčinou jsou různé mutace postihující gen pro LDL-receptor, který je umístěn na krátkém raménku 19. chromosomu. Postižení jedinci mají deficitní syntézu LDL-receptorů, nebo je tvoří normálně a porucha tkví v nemožnosti transportovat tyto receptory na povrch buňky, anebo je porušena vazba receptoru na lipoproteinovou částici; dále může váznout internalizace komplexu lipoprotein-receptor nebo uvolnění LDL z LDL-receptoru v endosomech a tím i recyklace LDL-receptorů (Obr. 14):
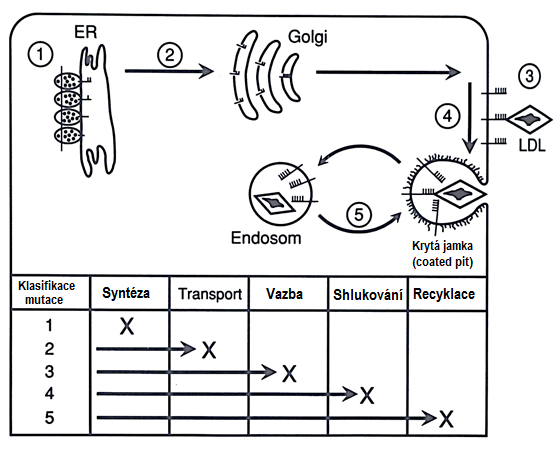
Obr. 14. Možné příčiny defektu LDL-receptoru (Goldstein a Brown 1984)
Základním rysem onemocnění jsou projevy předčasné aterosklerózy, zejména jako ischemická choroba srdeční (ICHS). Zatímco u heterozygotů se infarkt myokardu objeví počátkem čtyřicítky, homozygoti umírají bez léčby většinou na akutní infarkt myokardu do 20 let.
Hlavním znakem je hypercholesterolemie, přičemž biochemický nález bývá u heterozygotů 7–15 mmol/l, u homozygotů 16–23 mmol/l.
Z hlediska patobiochemie jde o deficit LDL-receptorů na povrchu fibroblastů, adipocytů a buněk hladké svaloviny (u homozygotů), případně o jejich snížení (u heterozygotů). LDL se neodbourávají normálním způsobem, hromadí se v cirkulaci a poškozují stěnu cév. LDL se neodbourávají pomocí regulovaných LDL-receptorů, ale jiným způsobem („scavenger“ cells – „zametací“ neboli „odklízecí“ buňky), LDL-cholesterol není internalizován v LDL‑receptorových buňkách. Tím nedochází k inhibici klíčového enzymu pro syntézu cholesterolu, kterým je hydroxymetylglutaryl-CoA-reduktasa (HMG-CoA-reduktasa); proto není potlačována syntéza cholesterolu v buňkách a je aktivována tvorba esterů cholesterolu, které se ukládají v intimě cévní stěny.
Ad 2) Familiární defekt apoproteinu B100 (odpovídá rovněž typu Fredrickson IIa)
Jde o genetický defekt v polypeptidu apolipoproteinu B-100. Bodovou mutací v poloze 3500 je zaměněn arginin za glutamin (odtud ApoB3500); tato změna molekuly apolipoproteinu B narušuje jeho schopnost vázat se na LDL-receptor. LDL částice se hromadí v plazmě, stoupá jak celkový cholesterol (7–10 mmol/l), tak především LDL-cholesterol a hladina ApoB. Diagnostika je založena na metodách molekulární biologie.
Ad 3) Poruchy PCSK9
Proprotein konvertasa subtilisin/kexin typ 9 (PCSK9) je enzym ze skupiny proproteinových konvertas, specifických serinových proteas, které se syntetizují především v játrech ve formě zymogenů. Přesný fyziologický význam této bílkoviny není známý. Tento enzym je syntetizovaný v endoplasmatickém retikulu ve formě neaktivní pro-PCSK9, která obsahuje prodoménu, katalytickou doménu a C-terminální doménu bohatou na cystein a histidin. V Golgiho komplexu je prodoména autokatalyticky odštěpena, ale zůstává nekovalentně navázána na zralou PCSK9, pomáhá při skládání tohoto enzymu a blokuje jeho katalytickou aktivitu. Jediným známým substrátem PCSK9 je tedy sám tento enzym.
Po rozštěpení se PCSK9 secernuje do krve a může se navázat na LDL-receptor na povrchu buněk. Jejím úkolem je posttranslační regulace počtu LDL-receptorů na povrchu buněk. Pro vazbu na LDL-receptor není katalytická aktivita PCSK9 potřeba. Komplexy LDL‑receptor‑PCSK9 jsou endocytosou internalizovány do buňky a posléze se rychle odbourávají v lyzosomech. LDL‑receptory nemohou být recirkulovány, namísto toho se odbourávají, takže zvýšení koncentrace PCSK9 vede k rychlému snížení dostupnosti tohoto receptoru.
U pacientů s familiární hypercholesterolemií byly popsány různé missence mutace v genu pro PCSK9, které vyvolávají její zvýšenou afinitu k LDL-receptoru nebo zvýšení koncentrace zralé PCSK9 díky změnám v intracelulárním zpracování proenzymu. Tyto změny způsobují výrazné snížení počtu LDL-receptorů a nárůst plasmatické koncentrace LDL‑cholesterolu.
B. Polygenní hypercholesterolemie
Hladina plasmatického cholesterolu je u tohoto typu hypercholesterolemie ovlivňována řadou faktorů, genetických i exogenních. Výskyt se odhaduje na 1 ze 100 až 200 osob. Kombinace několika nepříznivých genetických změn spolu s faktory zevního prostředí vede k obvykle mírnému zvýšení plasmatického cholesterolu (do 8 mmol/l), koncentrace HDL-cholesterolu a TAG bývají normální. Ateroskleróza je urychlena a riziko ICHS je tedy zvýšené.
C. Primární smíšené hyperlipidemie
a) Familiární kombinovaná hyperlipoproteinemie (odpovídá typu IIb podle Fredricksona)
Je nejčastější geneticky podmíněnou poruchou metabolismu lipoproteinů. Frekvence výskytu se odhaduje na 1:50-100, dědičnost je většinou autosomálně recesivní. Často se objevuje u obézních a u diabetiků. Patologické projevy z aterosklerózy (ICHS, ischemie dolních končetin) nastupují až v dospělosti. Z patobiochemického hlediska je za příčinu považována abnormálně vysoká syntéza Apo B v játrech, která je provázená zvýšenou produkcí VLDL.
b) Familiární dysbetalipoproteinemie (hyperlipoproteinemie typu III podle Fredricksona, zvýšení „β-VLDL“)
Tento typ je vzácný, jde o dědičnou chorobu charakterizovanou poruchou odstraňování zbytků chylomiker a VLDL. Podkladem této poruchy je homozygocie pro mutantní formu apo E (apoE2), které se špatně váže na jaterní receptory (viz výše, metabolismus lipoproteinů). V důsledku toho se hromadí remnanty chylomiker a také VLDL bohaté na cholesterol (někdy označované jako β-VLDL).
c) Primární hypertriacylglycerolemie (Familiární hyperchylomikronemie, familiární hyperlipoproteinemie typu I podle Fredricksona)
Tato vzácná choroba s autosomálně recesivní dědičností je způsobena chyběním lipoproteinové lipasy (LPL). Do stejné skupiny patří i defekt apoproteinu C-II, který lipasu aktivuje, nebo přítomnost inhibitoru LPL. Výsledkem defektu je hromadění chylomiker, která nemohou být normálně odbourávána a která jsou odstraněna pomocí makrofágů. U postižených není riziko aterosklerózy zvýšené, ale je značné riziko akutní pankreatitidy.
d) Familiární hypertriacylglycerolemie (typ IV podle Fredricksona)
Jde o poruchu v monogenní formě děděnou autosomálně dominantně. Projevuje se až v dospělosti a je poměrně častá, postihuje 0,2-0,3% populace. Zvýšení VLDL může být způsobeno zvýšením syntézy těchto částic v játrech, snad i snížením přeměn částic VLDL v cirkulaci.
e) Familiární zvýšení VLDL+chylomiker (hyperlipoproteinemie typ V podle Fredricksona)
Jde o onemocnění poměrně vzácné (1:5000), častěji se objevuje u dospělých, kteří jsou obézní, mají hyperurikemii a diabetes. Vyvolávajícím faktorem může být požívání alkoholu a léků s estrogeny nebo také renální insuficience.
Příčina vzniku tohoto typu není zcela vysvětlena. Soudí se, že jde o poruchu metabolismu VLDL a chylomikronů. Aktivita lipoproteinové lipasy je však normální (na rozdíl od Fredricksonova typu I). Příčina současného zvýšení VLDL a chylomikronů se vysvětluje třemi možnými způsoby: (1) Zvýšená tvorba a sekrece VLDL játry vede k saturaci mechanismu odstraňujícího částice bohaté na triacylglyceroly, tedy i chylomikrony, (2) Syntéza triacylglycerolů je normální, ale je porušen mechanismus jejich vychytávání, (3) Může jít o kombinaci obou mechanismů. Forma navozená požíváním alkoholu je v některých zemích (kupř. ve Francii) velmi častá. Rozlišení typu IV od V je obtížné, stejně jako odlišení od sekundární formy navozené dietou (nadměrný přívod tuků a sacharidů).
Hyperalfalipoproteinemie
Familiární hyper-α-lipoproteinemie
Jde o genetickou lipoproteinovou abnormalitu spojenou s výskytem dlouhověkosti v rodině (o 8-12 let oproti průměru v populaci); předpokládaná forma dědičnosti je autosomálně dominantní. Familiární formu je však nutno odlišit od formy získané (sekundární) kupř. při abúzu alkoholu nebo při užívání antikoncepčních preparátů nebo přípravků na bázi estrogenů.
Syndrom je charakterizován výrazným zvýšením HDL-cholesterolu (zvýšení a1-lipoproteinu při elektroforéze), mírné až střední zvýšení celkového cholesterolu v plasmě a normální koncentrace S-triacylglycerolů. Jsou zmnoženy HDL částice obsahující jen ApoAI nikoliv částice obsahující jak ApoAI, tak ApoAII [LpA I : A II]. Abnormalita je pravděpodobně způsobena zvýšenou syntézou apo AI. Je snížené riziko kardiovaskulárních chorob navozených aterosklerózou.
Hypolipoproteinemie
Familiární hypo-β-lipoproteinemie
Je považována zatím za vzácnou genetickou abnormitu, pravděpodobně s autosomálně dominantní dědičností. Hladina LDL cholesterolu v plasmě je snížena. Podobně jako vzácná hyper-α-lipoproteinemie je i tato anomálie sdružena s dlouhověkostí, pravděpodobně pro nízkou incidenci infarktů myokardu.
Abetalipoproteinemie
Jde o vzácnou autosomálně recesivně přenášenou chorobu, při níž není syntetizován Apo‑B48 v enterocytech ani apo-B100 v játrech. Proto chybí v cirkulaci chylomikrony a je narušen transport endogenního cholesterolu k periferním buňkám cestou LDL a koncentrace cholesterolu v plasmě je snížena. Zhoršené vstřebávání lipidů ze střeva způsobí jejich nadměrné ztráty stolicí a tím i nedostatek vitaminů rozpustných v tucích. Heterozygoti nemají žádné zjevné klinické příznaky, u homozygotů se popisují pestré příznaky, mj. zpožděný vývoj a snížený intelekt, polyneuropatie a svalová slabost.
Hypoalfalipoproteinemie
Stavy se sníženým HDL-cholesterolem mají zvýšené riziko aterosklerózy a z ní plynoucí kardiovaskulární onemocnění. Familiární forma se zdá mít dominantní dědičnost. Byly popsány i abnormality v polypeptidové složení apoA-I a jeden takovýto lipoprotein byl nazván podle místa popsaného případu apoA-I-Milano. Pacienti jsou po většině asymptomatičtí. Bez apoA-I se nemůže tvořit HDL a bez HDL nemůže být apoC-II transportován zpět do jater v průběhu odbourávání VLDL. Důsledkem je relativní nedostatek apoC-II a zvýšená hladina VLDL.
Analfalipoproteinemie (Tangierská nemoc)
Je to vzácná choroba s autosomálně recesivní dědičností, charakterizovaná úplným chyběním HDL v plasmě. Homozygoti mají nedetekovatelné množství HDL-cholesterolu a extrémně nízký apoA-I a apoA-II. Na elektroforéze lipoproteinů chybí a-frakce. Celkový cholesterol i LDL-cholesterol jsou sníženy; je mírná hypertriacylglycerolemie. Biochemický defekt tkví pravděpodobně v abnormálně rychlém katabolismu HDL a apoA-I.
Poznámka:
Polékové dyslipidemie
Existuje řada léčiv, která zasahují do metabolismu plasmatických lipidů. Významné jsou samozřejmě ty lékové skupiny, které jsou často součástí medikace nemocných s dyslipidemií. Je popisován vliv podávání beta-blokátorů na hladiny krevních lipidů. Nemocní s ischemickou chorobou srdeční mají tuto lékovou skupinu ve svých medikacích pravidelně a stejně pravidelně je u nich zjišťována dyslipidemie. Při jejich podávání se objevuje zvýšení triacylglycerolů a snížení HDL, což se dává do souvislosti se snížením aktivity lipoproteinové lipasy nebo beta‑blokátory navozenou redistribucí krve se zmenšením průtoku kosterním svalstvem, kde je vysoká koncentrace tohoto klíčového enzymu. Jinou skupinou jsou orální kontraceptiva. Estrogeny mají schopnost zvýšit produkci částic VLDL hepatocytem, ale zároveň zvyšují jejich odbourávání, takže celková koncentrace triacylglycerolů se nemění. U disponovaných žen se po zahájení podávání kontraceptiv může objevit zvýšení triacylglycerolů, bývá vyšší frakce chylomiker.