Lipidózy
Lipidózy patří do skupiny lyzosomálních chorob, což jsou vzácná, dědičně podmíněná onemocnění, charakteristická hromaděním nefunkčních meziproduktů (střádavé choroby).
Lyzosomy jsou bohaté na hydrolyzující enzymy. Tzv. kyselé hydrolasy umožňují postupné štěpení velkých komplexních molekul (lipidů, glykoproteinů, mukopolysacharidů) z buněčných stěn zanikajících buněk. Nedostatečné množství či nedostatečná enzymatická aktivita vede k hromadění substrátů těchto defektních enzymů v lyzosomech postižených buněk. Vedle defektů hydrolas mohou být příčinou lyzosomálních chorob např. poruchy transportu proteinů do lyzosomů, deficit lyzosomálních membránových proteinů nebo deficit aktivátorů lyzosomálních hydrolas.
Lyzosomální choroby jsou multisystémová onemocnění s trvalou progresí, která se mohou manifestovat kdykoli v průběhu života. Postižené jsou především metabolicky aktivní orgány a tkáně (kostní dřeň, kosti, kosterní svaly, myokard a CNS). Bylo popsáno cca 60 různých lyzosomálních poruch. Časné formy mívají těžký průběh s rychlou progresí a infaustní prognózou. Výskyt v naší populaci se odhaduje na 1:8200 živě narozených dětí. Většinou se jedná o autosomálně recesivně dědičné choroby, vzácně gonosomálně recesivní (Fabryho nemoc a mukopolysacharidóza typu II).
Výraznou skupinou lyzosomálních chorob jsou sfingolipidózy, charakteristické strukturou látek a tkáňovou lokalizací hromadění meziproduktů.
Sfingolipidy
Sfingolipidy tvoří rozsáhlou skupinu lipidů, které ve své molekule obsahují alifatické nenasycené aminoalkoholy, tzv. sfingosiny. Základem struktury všech sfingolipidů je ceramid (N-acylsfingosin), který vzniká substitucí aminoalkoholu sfingosinu na C2 mastnou kyselinou. Substitucí polárním substituentem na C1 ceramidu vznikají fosfosfingolipidy (též sfingomyeliny) a glykosfingolipidy (cerebrosidy, gangliodidy, globosidy, sulfatidy) (Obr. 12).
Sfingomyeliny vznikají esterifikací primární alkoholové skupiny sfingosinu kyselinou fosforečnou, jejíž další hydroxylová skupina je esterifikovaná cholinem. Jedná se o nejzozšířenější sfingolipidy v živočišných tkáních. Nacházejí se v bílé hmotě mozkové a v myelinových pochvách nervových vláken.
V molekulách glykosfingolipidů je k primární alkoholové skupině sfingosinu O‑glykosidickou vazbou připojena cukerná složka. Pokud se jedná o jednu hexosu (nejčastěji glukosu, galaktosu, nebo jejich deriváty) hovoříme o cerebrosidech. Ty tvoří strukturní základ složitějších glykosfingolipidů, jako jsou globosidy (přítomné v krevní plasmě a erythrocytech), sulfatidy (vyskytují se v mozku a ledvinách) a gangliosidy.
Gangliosidy obsahují kromě několika molekul monosacharidových jednotek i některou sialovou kyselinu, což jsou acylované deriváty neuraminové kyseliny. V lidských gangliosidech se vyskytuje kyselina N-acetylneuraminová (NANA). Tyto látky mají odlišné složení mastných kyselin než ostatní glykosfingolipidy, vyskytuje se v nich především kyselina stearová (Obr. 13). Vyskytují se v šedé kůře mozkové a slezině a podílejí se na rozpoznávání buněk.
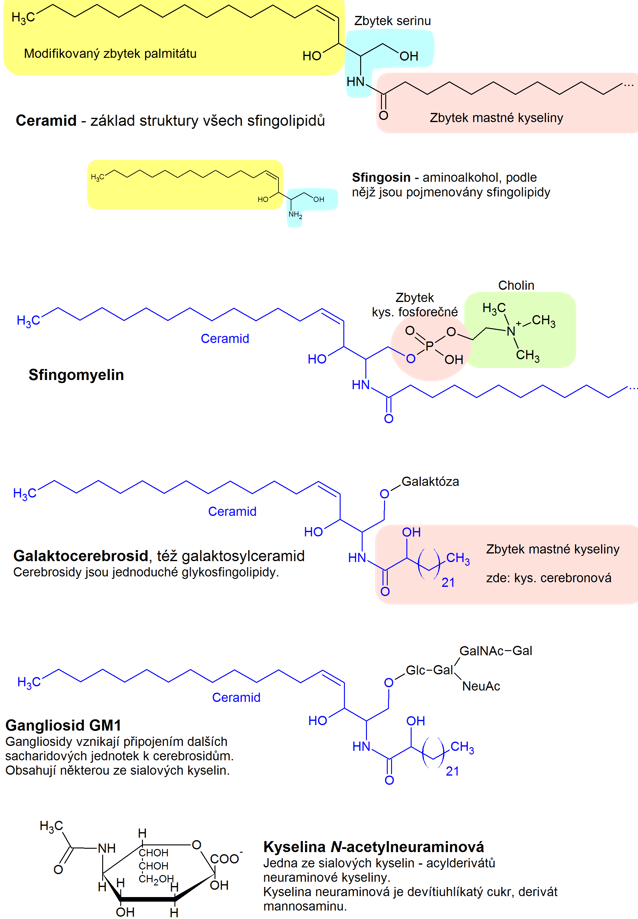
Obr. 12. Struktura sfingolipidů (Vejražka 2017)
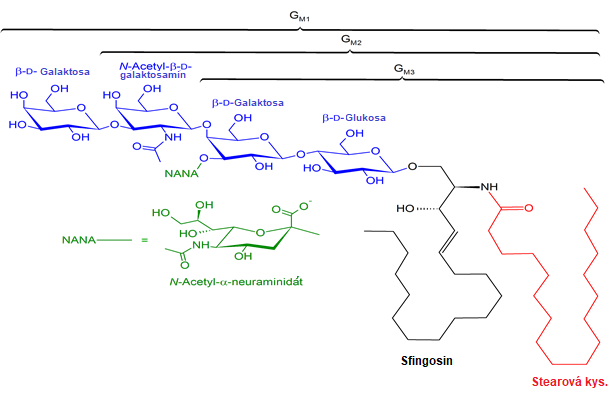
Obr. 13. Struktura gangliosidů GM1, GM2, GM3 (Anonymní autor 2012)
Sfingolipidózy
Sfingolipidózy jsou dědičné choroby, způsobené genetickou mutací některého enzymu s následkem hromadění meziproduktu, na němž se štěpení zastavuje (Obr. 14). Postižen je zejména nervový systém, v němž je účast sfingolipidů v membránách charakteristická. Z hlediska patologie je důležité, že nemetabolizovatelné meziprodukty se hromadí. V centrálním nervovém systému je problém mj. v tom, že prostor je omezen lebeční dutinou, takže hromadění meziproduktů zároveň představuje významný útlak funkčních struktur. Odtud se odvíjí postižení funkce CNS, vč. mentálních poruch. U postižení periferních nervů se střádáním nemetabolizovatelných meziproduktů se nápadně objevuje neuroviscerální symptomatologie a také zvětšování parenchymatózních orgánů (hepatomegalie, splenomegalie, hepatosplenomegalie).
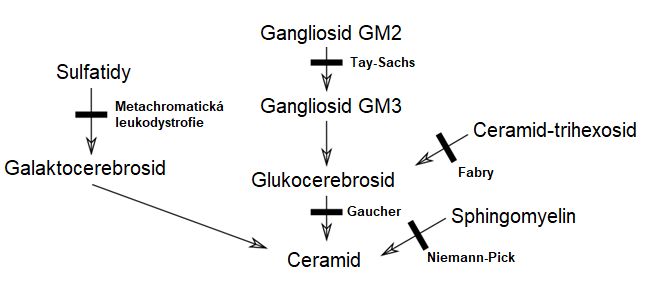
Obr. 14. Schéma defektů štěpení sfingolipidů (Anonymní autor 2010)
Příklady sfingolipidóz s klinickými příznaky uvádí tabulka 4.
Tabulka 4. Příklady sfingolipidóz (Murray et al. 2012)
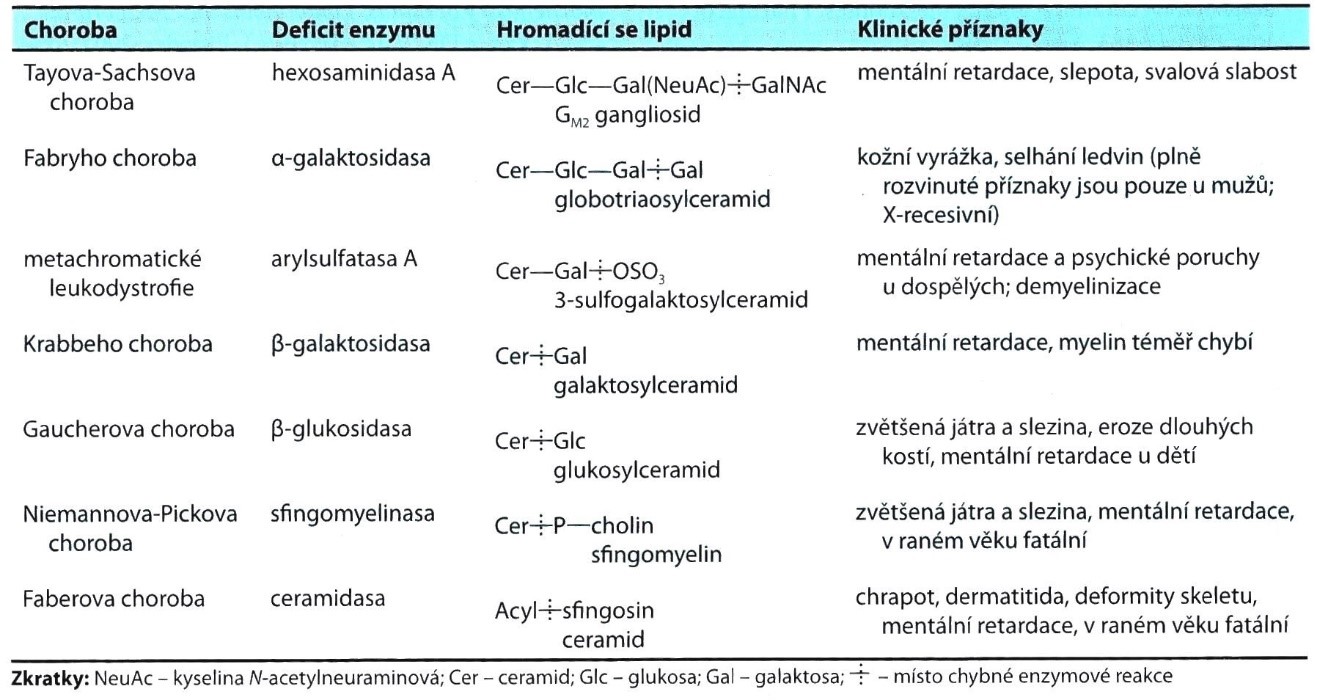
Některé sfingolipidózy lze léčit substitučním podáváním rekombinantních enzymů (např. Gaucherova nebo Fabryho choroba), nebo dokonce omezením množství střádaného substrátu („substrát redukční terapie“ u Gaucherovy nemoci).