Buněčné a molekulární základy nádorového onemocnění
Díky různorodým genomickým a epigenomickým změnám získávají nádorové buňky během karcinogeneze jedinečné fenotypové vlastnosti. Všechny nádory se projevují osmi základními změnami v buněčné fyziologii, které jsou považovány za typické znaky nádoru (Obr. 2). Typické znaky nádorových buněk zahrnují: 1) soběstačnost v produkci růstových signálů (aktivace onkogenů), 2) necitlivost k signálům zastavujícím buněčný cyklus (inaktivace tumor-supresorových genů), 3) schopnost přeprogramovat energetický metabolismus buňky (Warburgův efekt – posílení aerobní glykolýzy a potlačení mitochondriálního metabolismu), 4) schopnost obejít apoptosu, 5) nepřetržitá tvorba nových kapilár (angiogeneze), 6) schopnost invadovat a tvořit metastázy, 7) neomezený replikační potenciál (nesmrtelnost) a 8) schopnost uniknout před imunitní odpovědí hostitele. Získání genetických a epigenetických alterací, které tyto charakteristické rysy propůjčují, může být urychleno nestabilitou genomu nádorových buněk a zánětem podporujícím nádorové bujení. Ty jsou považovány za umožňující charakteristiky, protože podporují buněčnou transformaci a následnou progresi nádoru. Ovšem vlastní průběh karcinogeneze je individuální a pořadí i počet získaných vlastností se u jednotlivých nádorů liší, stejně tak se nádory liší i konkrétními zasaženými geny.
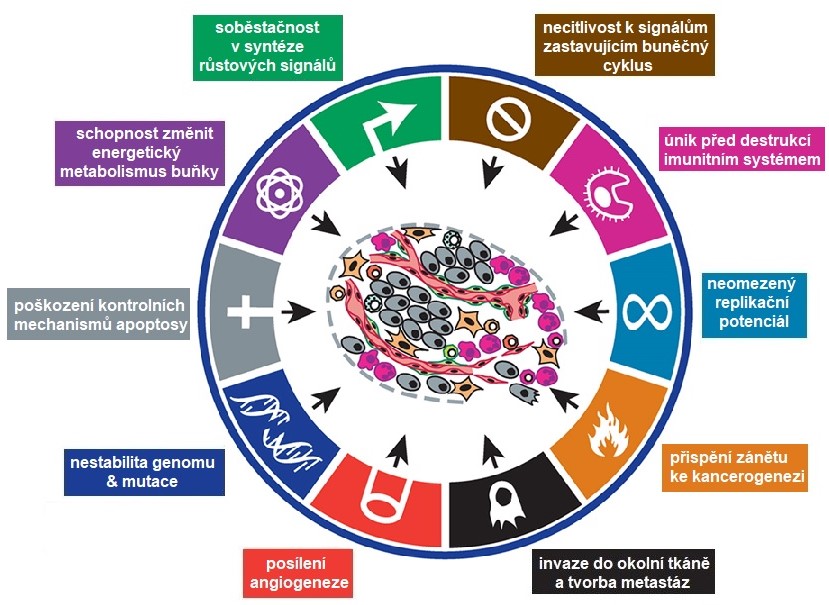
Obr. 2. Charakteristické rysy nádorových buněk (upraveno z Hanahan a Weinberg 2011)
Maligní nádor vzniká na podkladě kumulace somatických alterací určitých genů vyvolaných genotoxickým působením fyzikálních, chemických nebo biologických vlivů. Vznik maligního nádoru u postiženého jedince může být urychlen existencí hereditárních mutací. Na jeho rozvoj mají významný dopad i epigenetické faktory ovlivňující expresi genetické informace. U solidních nádorů dochází v průběhu karcinogeneze k nahromadění průměrně šesti až osmi kritických mutací, tedy mutací, které jsou přímým podkladem maligní transformace nádorových buněk. Mezi kritické mutace patří aktivace protoonkogenů, inaktivace anti-onkogenů (tumor-supresorových genů), zvýšení exprese inhibitorů apoptosy a mutace genů řídících průběh buněčného cyklu.
Protoonkogeny a jejich aktivace
Protoonkogeny jsou vysoce konzervované normální geny přítomné v genomu všech buněk. Jejich produkty se podílejí na regulaci buněčné proliferace, diferenciace a apoptosy normálních buněk. Působí jako pozitivní regulátory buněčného dělení (stimulace dělení) a negativní regulátory apoptosy (inhibice apoptosy). Protoonkogeny snadno podléhají genetickým změnám, které umožní jejich aktivaci. Pokud jsou aktivovány nebo modifikovány na strukturní nebo kontrolní úrovni, začnou se chovat jako onkogeny a podporují vývoj nádoru. V současnosti je známo asi 100 onkogenů. Mutace, které vedou ke změně protoonkogenů na onkogeny, jsou aktivující a dominantní (stačí změna jedné alely).
Onkogeny byly objeveny díky retrovirům nesoucím virové analogy buněčných onkogenů. Tyto virové onkogeny byly objeveny při studiu genomu těchto virů dříve než onkogeny buněčné, což podporovalo představu o obecném virovém původu nádorů. Technikami hybridizace se však zjistilo, že normální buňky mají identické nebo podobné sekvence jako tyto virové onkogeny. Ty jsou tedy odvozeny od lidských (obecně eukaryontních) „protoonkogenů“, které byly přetaženy z hostitelské buňky a zabudovány do genomu retroviru v průběhu jeho životního cyklu.
Názvy jako onkogen a protoonkogen sugerují představu, že funkcí těchto genů je navodit nádorovou transformaci buňky. Teprve později se zjistilo, že protoonkogeny jsou významné geny, jejichž produkty představují molekuly, účastnící se řádných buněčných dějů, jakými jsou diferenciace, regulace, signalizace nebo recepce signálů.
Nomenklatura těchto genů je většinou odvozena od primárně zjištěného účinku virového onkogenu, vžila se a používá se i nyní, kdy je už známa normální buněčná funkce příslušné molekuly. Např. názvy ras pro gen a ras pro příslušný protein jsou odvozeny od toho, že tyto geny byly objeveny v retrovirech, způsobujících sarkom u potkanů: rat sarcoma Þ ras. Podobně sis od simian sarcoma (opičí sarkom), fes od feline sarcoma (kočičí sarkom) atp. (podrobnější informace jsou uvedeny dále v textu). V kontextu virové transformace pak označujeme buněčný protoonkogen jako c-onc a jeho virovou formu jako v‑onc (např. v-ras a c-ras).
Protoonkogeny regulují replikaci DNA a přenosy signálu přes své produkty - proteiny. Protoonkogeny můžeme dělit podle funkce jejich produktu na růstové faktory, receptory pro růstové faktory, cytoplasmatické proteiny (proteinkinasy a G-proteiny), jaderné proteiny. Známé produkty protoonkogenů a onkogenů spadají do několika funkčních skupin a jsou označovány třemi písmeny: růstové faktory (sis, hst), receptory pro růstové faktory (erbB, fms, kit) a tyrosinkinasy (src, fgr, fcs, trk), membránově-vázané analogy proteinů vážících GTP (gsp, gip, ras) cytoplasmatické serinové a threoninové kinasy (mil, mos, raf), jaderné proteiny fungující jako aktivátory transkripce (myb, myc, fos, jun). U řady lidských nádorů byla prokázána zvýšená exprese členů jednotlivých tříd, která pak přispívá k řadě vlastností nádorů (např. buněčná proliferace, invazivita a tvorba metastáz).
Způsoby aktivace a změny protoonkogenu na onkogen jsou uvedeny v Obr. 3. Jejich výsledkem je narušení struktury protoonkogenu, nebo zvýšení jejich exprese. Mechanismy aktivace protoonkogenů zahrnují mutace, genové amplifikace a chromosomální přestavby. Malá změna DNA (bodová mutace nebo delece) v kódující oblasti genu může způsobit vznik proteinu se zvýšenou aktivitou, zatímco tato změna v regulatorní oblasti daného genu může vyvolat nadprodukci normálního proteinu. Chyby v replikaci DNA mohou být příčinou zvýšení počtu kopií genu (amplifikace), což může vést k redundantní replikaci DNA a nadbytečné expresi (nadprodukci) normálního proteinu. Chromosomální přestavba (translokace) v kódující sekvenci genu může vyvolat fúzi protoonkogenu s jiným genem, jejímž výsledkem bude vznik fúzního (chimerního) proteinu se zvýšenou aktivitou (např. Filadelfský chromosom u chronické myeloidní leukemie). Naproti tomu alterace v regulatorní oblasti genu, jako je přemístění genu k vysoce aktivní promotorové sekvenci (např. poblíž genů pro imunoglobuliny nebo T-receptory), mohou způsobit, že gen bude přepisován mnohem častěji než za normálních podmínek (např. Burkittův lymfom). Mutace v buněčném genomu vedoucí k maligní transformaci mohou být způsobeny také onkogenními viry (nejčastěji retroviry, viz dále).
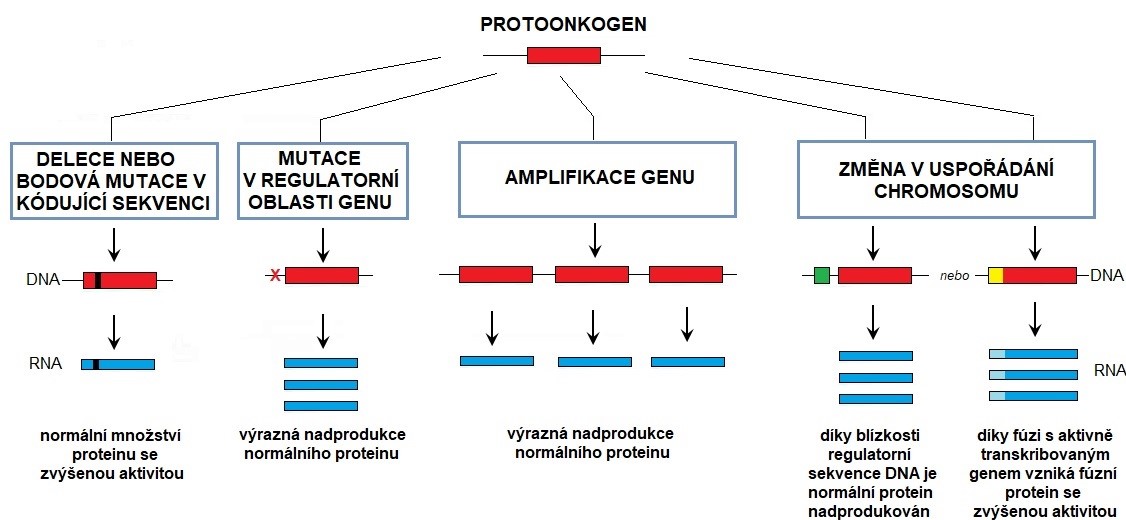
Obr. 3. Způsoby aktivace a změny protoonkogenu na onkogen (upraveno z Alberts a kol. 2007)
Jako příklady chromosomálních aberací, které jsou příčinou aktivace protoonkogenu, uvádíme translokaci genu c-myc (Burkittův lymfom) a vznik Filadelfského chromosomu (chronická myeloidní leukemie). U Burkittova lymfomu dochází k chromosomové přestavbě a to sice k translokaci mezi osmým a čtrnáctým chromosomem t(8;14), která je vyvolána virem Epsteina a Barrové. Buněčný protoonkogen c-myc je přemístěn z 8. chromosomu na chromosom 14, kde se dostává do blízkosti aktivního promotoru genu pro těžký imunoglobulinový řetězec. Následně dochází k neregulované expresi genu c-myc, který kóduje transkripční faktor C-MYC podílející se na regulaci buněčného cyklu, apoptosy a buněčné transformace (Obr. 4A). V případě Filadelfského chromosomu, který je typický pro myeloidní leukemie, dochází k translokaci protoonkogenu abl z 9. chromosomu na chromosom 22 t(9;22). Protoonkogen abl se při translokaci připojí uprostřed genu bcr, který má silný promotor. Vzniká fúzní gen bcr-abl, který po přepisu poskytuje fúzní protein BCR-ABL. Produkt genu abl na původní pozici fyziologicky stimuluje buněčnou proliferaci (má tyrosinkinasovou aktivitu), zatímco fúzní protein BCR-ABL stimuluje buňky k nekontrolovatelnému dělení (Obr. 4B).
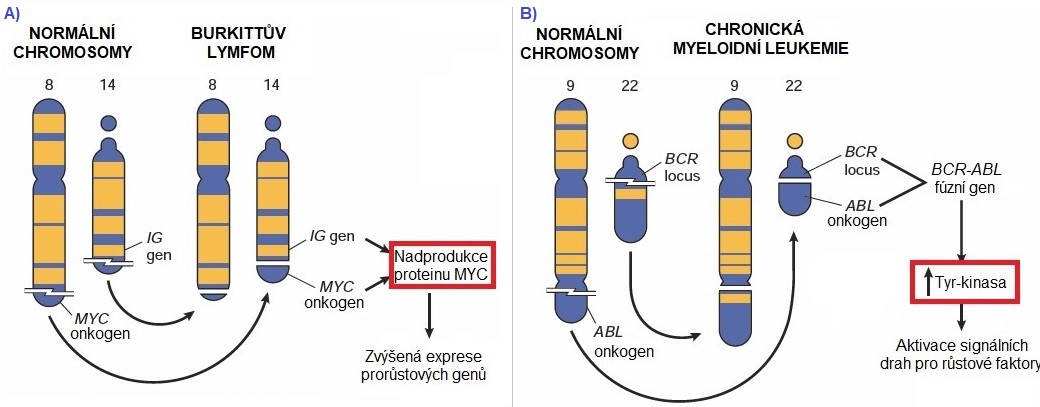
Obr. 4. Chromosomální translokace u Burkittova lymfomu a chronické myeloidní leukemie (upraveno z Kumar a kol. 2014)
Tumor-supresorové geny
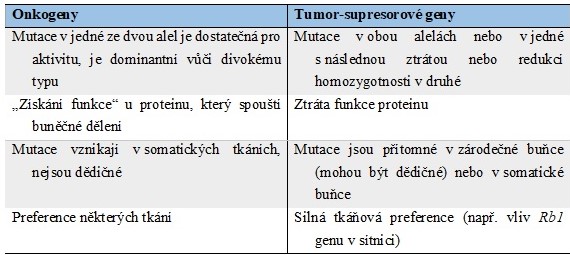
Buněčné dělení je kromě pozitivní stimulace růstovými faktory či hormony řízeno rovněž negativní regulací. Tuto negativní regulaci zprostředkovávají produkty tumor-supresorových genů (antionkogenů), které potlačují proliferaci buněk, působí jako kontrolní body buněčné proliferace nebo programované buněčné smrti. Tyto proteiny tedy fungují jako negativní regulátory buněčného cyklu (antiproliferační signály, inhibice mitosy a buněčného růstu) a pozitivní regulátory apoptosy (navození buněčné smrti). Odhaduje se, že v lidském genomu je přítomno asi 30 tumor-supresorových genů. Transkripce tumor-supresorových genů je aktivována při buněčném stresu nebo poškození, aby se zabránilo abnormální proliferaci a přenosu poškozené genetické informace. Mutace, delece nebo epigenetická regulace (např. methylací DNA) těchto genů umožňuje proliferaci postižené buňky a výrazně zvyšuje riziko vzniku nádoru. Mutace zasahující tumor-supresorové geny jsou inaktivující a recesivní (nutná změna obou alel – teorie dvojího zásahu). Rozdíly mezi onkogeny a tumor-supresorovými geny jsou shrnuty v Tabulce 1.
Tab. 1. Některé rozdíly mezi onkogeny a tumor-supresorovými geny (upraveno z Rodwell a kol. 2015)
Mezi známé tumor-supresorové geny patří gen Rb, APC, p53 nebo BRCA 1/2. Předpokládá se, že v některých případech je to právě ztráta funkce genů kódujících tyto proteiny, co způsobuje vývoj nádoru. Prvním objeveným tumor-supresorovým genem byl retinoblastomový gen Rb, který je zapojen do regulace buněčného cyklu. Produkt tohoto genu protein pRb váže transkripční faktory E2F, které jsou nutné při přechodu buňky z G1 do S fáze. Cyklin-dependentní kinasy fosforylují a tím inaktivují pRB, po fosforylaci se totiž změní konformace pRb a dojde k uvolnění faktoru E2F, který stimuluje transkripci genů nutných pro postup buňky do S fáze (např. cykliny A a E a enzymy replikace DNA). V případě mutace genu Rb nebo při sekvestraci pRb proteiny adenovirů či papilomavirů dojde ke vzniku maligního nádoru sítnice – retinoblastomu.
Protein p53 („strážce genomu“) je transkripční faktor, který je aktivovaný při poškození DNA, a brání růstu a rozvoji nádorů. Tento protein je klíčový pro udržení genetické stability buňky, protože omezuje poškození genomu. V normálních buňkách je exprese p53 velmi nízká a zvyšuje se při oxidačním stresu a po ozáření UV světlem. Při poškození DNA navozuje protein p53 v jádře zvýšení transkripce genu pro protein p21, který inhibuje cyklin-dependentní kinasy (CDK) a dočasně tak zastavuje buněčný cyklu v G1 fázi. Následně protein p53 indukuje opravy DNA prostřednictvím stresových proteinů GADD45[1] a pokud je DNA neopravitelně poškozená, pak indukuje apoptosu buňky cestou regulátoru apoptosy BAX[2]. Nefunkční p53 způsobí, že poškozené/mutované buňky pokračují v buněčném cyklu, umožní poškozeným buňkám vyhnout se apoptose a umožní vznik genetické nestability s následnou akumulací mutací (Obr. 5). Mnohostrannost p53 naznačuje, že jeho mutace je zdrojem řady poruch. Mutace p53 patří mezi nejčastější genetické změny v lidských nádorech a byly popsány až u poloviny všech lidských nádorů. Pokud jedinec zdědí od rodičů pouze jednu funkční alelu genu p53, je predisponován k rozvoji nádorového onemocnění a obvykle se u něj v dětství či v časné dospělosti vyvine několik zhoubných nádorů v různých tkáních (např. osteosarkomy a sarkomy měkkých tkání, karcinom prsu, adenokarcinom dřeně nadledvin, nádory CNS, leukémie). Toto onemocnění se označuje jako Li-Fraumeniho syndrom.
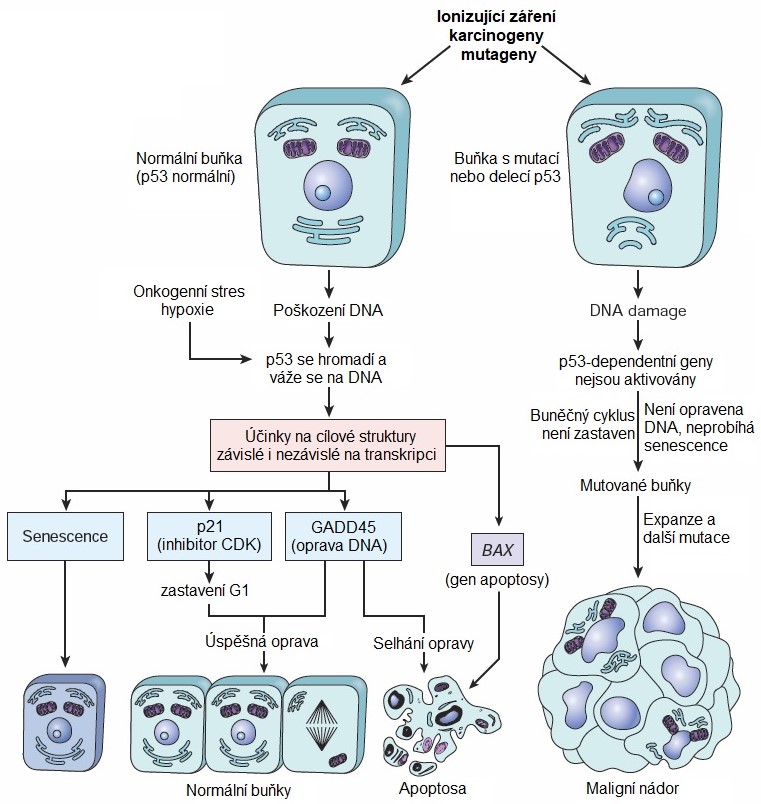
Obr. 5. Úloha p53 v zachování integrity genomu (upraveno z Kumar a kol. 2014).
Poruchy reparačních genů
Na vzniku nádorů se podílejí rovněž poruchy reparačních genů, které zabezpečují zachování genetické stability, které je pro přežití organismu nezbytné. Genetická stabilita je zajištěna velmi přesnou replikací DNA a reparací případných mutací. Opravné procesy poškozených bazí, abazických segmentů či jednořetězcových zlomů se významně liší od opravných procesů dvouřetězcových zlomů DNA. Integrita DNA je v průběhu buněčného cyklu pravidelně kontrolována v klíčových kontrolních bodech („checkpoint“) mezi fázemi G1 a S, v průběhu syntézy DNA, po dokončení syntézy DNA mezi fázemi S a G2 a mezi fázemi G2 a M. V případě poškození DNA zastavují produkty reparačních genů buněčný cyklus a dávají buňce možnost opravy chyb. Mezi opravné geny patří geny kódující různé proteinkinasy, např. „checkpoint“ kinasy 1 a 2, jejichž aktivace zastavuje buněčný cyklus v bodech G1-S a G2‑M, nebo kinasy ATM a ATR[3], které se uplatňují v reparaci dvouřetězcových zlomů DNA. Mutace opravných genů samy o sobě neposkytují buňce možnost nekontrolované proliferace, ale zvyšují frekvenci výskytu mutovaných protoonkogenů a tumor-supresorových genů.
Telomery a telomerasy - imortalizace buněk
Normální somatické buňky mají omezený počet dělení a tedy i délku života. U lidských somatických buněk je maximální počet dělení (tzv. Hayflickův limit) kolem 60-70, po jeho překročení se již buňka nemůže dělit, nastává ireverzibilní zástava růstu a buňka vstoupí do období senescence, které může vyústit v apoptózu. Senescentní buňka se sice nemůže dělit, ale zůstává metabolicky aktivní. Podstatou Hayflickova limitu je zejm. postupné zkrácení telomer pod kritickou délku, která již neumožňuje další replikaci chromozomu.
Telomery jsou koncové části jaderných chromosomů eukaryot. Jedná se o vysoce konzervované nukleoproteinové komplexy, které obsahují tandemově se opakující sekvence DNA bohaté na guanin (TTAGGG) obalené specifickými proteiny, které telomery chrání před degradací. Telomery jsou klíčovým stabilizačním faktorem koncových částí chromosomu, umožňují rozpoznat přirozené konce chromosomů od míst s dvouřetězcovými zlomy, chrání DNA před působením degradačních enzymů (např. DNA-exonukleas) a uplatňují se při replikaci DNA.
Replikace DNA vyžaduje pro tvorbu nového vlákna DNA-polymerasu, deoxyribonukleotidy a primer, což je oligonukleotidová sekvence RNA, která se na základě komplementarity bazí připojuje k vláknu DNA a umožňuje zahájit jeho replikaci. U vyšších živočichů nemůže DNA‑polymerasa sama iniciovat syntézu nového vlákna DNA a iniciaci tedy zajišťuje navázání na primer. Po ukončení replikace je primer odstraněn. Vzhledem k tomu, že DNA-polymerasa umí přidávat nové nukleotidy pouze na 3' konec nově vznikajícího řetězce DNA, nemůže DNA-polymerasa po odstranění primeru na 5' konci dceřiného vlákna nahradit odstraněné sekvence nukleotidů primeru, takže dceřiné vlákno DNA je na 5' konci po každé replikaci kratší. Proto jsou na koncích chromosomů přidány repetitivní nekódující sekvence – telomery, na nichž se odehrává zkracování DNA po replikaci. Během každé replikace dojde ke zkrácení o 50 – 200 párů bazí. Když dosáhnou telomery kritické délky, je odkrytý konec chromosomu buněčnými mechanismy vyhodnocen jako dvouřetězcový zlom a dojde k aktivaci kontrolních bodů pRB a p53, které zastaví proliferaci (tzv. fáze mitotické krize). Buňka přechází do senescence a může být aktivována apoptosa.
Tento mechanismus je u nádorových buněk narušen aktivací genu kódujícího enzym telomerasu. Jedná se o reverzní transkriptasu, která rozšiřuje telomerické TTAGGG opakované sekvence na konci chromosomů, čímž vzniká templát pro dosyntetizování opožďujícího se řetězce. Telomerasa tak blokuje stárnutí a umožňuje buňce neomezeně se dělit (imortalizace). Většina lidských buněk nemá aktivní telomerasu (gen je však přítomný v genomu všech buněk), výjimkou jsou zárodečné, embryonální a hematopoetické buňky. Nádorové buňky jsou schopné obnovit aktivitu telomerasy, což jim umožní neomezeně se dělit.
Imortalizované buňky vznikají ve stadiu mitotické krize. Příčinou bývá inaktivace p53 a pRB, zvýšená exprese onkogenů c-Myc a Ras a vážná poruchy genomové stability. Ve stadiu krize dochází k aktivaci dráhy, která spojí odhalené konce dvou nehomologních chromosomů za vzniku fúzního dicentrického chromosomu, který je v další anafázi rozštěpen a tak vzniknou další dvouřetězcové zlomy DNA. Při každém dalším dělení se poškození genomu zvětšuje díky cyklu zlom-fúze-přemostění, buňka se tak ocitá na prahu mitotické katastrofy a buněčné smrti. Pokud je buňka schopná reaktivovat telomerasu, může pak prodloužit své telomery, obnovit chromosomální stabilitu a přežít. Takové buňky však pravděpodobně utrpěly poškození onkogenů a tumor-supresorových genů a je u nich značné riziko maligní transformace. Prakticky u všech typů nádorů jsou telomery zachovány a v 85-95 % případů je to díky upregulaci telomerasy. Zbývající nádory používají k udržení délky telomer mechanismus ALT (alternativní prodloužení telomer), který pravděpodobně závisí na rekombinaci DNA.
Apoptosa
K akumulaci neoplastických buněk může docházet nejen díky aktivaci onkogenů nebo inaktivaci tumor-supresorových genů, ale i díky mutacím v genech regulujících apoptosu. Apoptosa je jedním z mechanismů programované buněčné smrti, dalšími typy jsou např. autofagie, anoikis, nekroptosa či kornifikace. Jedná se o fyziologický proces, který slouží k odstranění nepotřebných nebo poškozených buněk. Buňka umírající apoptoticky vykazuje charakteristické morfologické znaky (např. kondenzace a fragmentace chromatinu, tvorba výběžků cytoplazmatické membrány, celkové zmenšení buňky a nakonec rozpad buňky na tzv. apoptotická tělíska), které ji
odlišují od nekrotických buněk. V apoptotické buňce jsou aktivovány cysteinové proteasy – kaspasy, které štěpí řadu cílových proteinů, a nukleasy, degradující DNA. Kaspasy se dělí na iniciátorové a efektorové. Iniciátorové kaspasy (CASP-8, -9, -10) interagují s proteiny spouštějícími apoptosu, aktivací těchto kaspas dojde ke spuštění efektorových kaspas (CASP-3, -6, -7) a programované buněčné smrti.
V buňce existují dvě základní dráhy (vnitřní a vnější), jejichž spuštění vede k aktivaci kaspas a apoptose. Vnitřní (mitochondriální) dráha je aktivována celou řadou intracelulárních podnětů (např. poškození DNA, oxidační stres, přetížení buňky Ca2+, nahromadění chybně složených proteinů v ER, nedostatek signálů pro přežití atd.). Do její regulace se zapojuje celá řada proapoptotických (p53 a BAX) i anti-apoptotických proteinů (rodina Bcl-2). Pokud převáží proapoptotické signály, dojde k permeabilizaci vnější mitochondriální membrány (MOMP) a následnému úniku cytochromu c z mezimembránového prostoru do cytoplasmy, který spolu s proteinem Apaf-1 aktivuje proCASP-9 na aktivní CASP-9. Z mitochondrií jsou dále uvolňovány proteiny Smac/Diablo, Arts či proteasa Omi/HtrA2, které vážou a inaktivují anti-apoptotické proteiny IAP („inhibitors of apoptosis“) přítomné v cytoplasmě. Vnější (receptorová) dráha je aktivována vazbou extracelulárních ligandů smrti (TRAIL, FasL, TNF[4]) na membránové receptory smrti (DR4/ DR5, Fas, TNFR[5] a další), které přes další adaptorové proteiny aktivují proCASP-8 a proCASP-10. Obě dráhy posléze konvergují do společné apoptotické dráhy, která začíná aktivací efektorových kaspas. Aktivované iniciátorové CASP-8, -9 a -10 aktivují efektorové kaspasy (CASP-3, -6 a -7) zajišťující inaktivační či aktivační štěpení řady proteinů, jako jsou regulátory apoptosy, proteiny související s buněčnou adhezí, cytoskeletem, strukturou jádra, buněčným cyklem, opravou a syntézou DNA, což nakonec vyústí v eliminaci buňky poškozené nebo z jiných důvodů určené k likvidaci (Obr. 6).
V nádorových buňkách je mutována celá řada proteinů, jež přímo či nepřímo ovlivňují rezistenci buněk k indukci apoptosy. Byly detekovány mutace proteinů, které jsou přímou součástí jak vnější dráhy (vzácněji), tak vnitřní dráhy od mitochondriálních regulátorů po kaspasy. Mezi mechanismy, které nádorovým buňkám umožňují uniknout buněčné smrti, patří mutace/ztráta p53, snížené vyplavení cytochromu c díky upregulaci anti-apoptotických faktorů (např. Bcl-2), ztráta Apaf1, upregulace inhibitorů apoptosy IAP, snížené množství receptoru smrti CD95 a inaktivace „death-induced signaling complex“. Dobře popsaná je úloha Bcl-2 v protekci maligních lymfoidních buněk před apoptosou. U většiny hematopoetických a solidních tumorů je nadměrně exprimován alespoň jeden člen anti-apoptotických proteinů rodiny Bcl-2 (díky translokaci genu do blízkosti silného promotoru imunoglobulinu), což naznačuje, že únik před apoptosou má velký význam v rozvoji a progresi nádorových onemocnění. Nadměrná exprese členů rodiny Bcl-2 je významným faktorem rezistence nádorů k terapii, protože při chemo- i radioterapii jsou nádorové buňky usmrcovány právě díky indukci apoptosy.
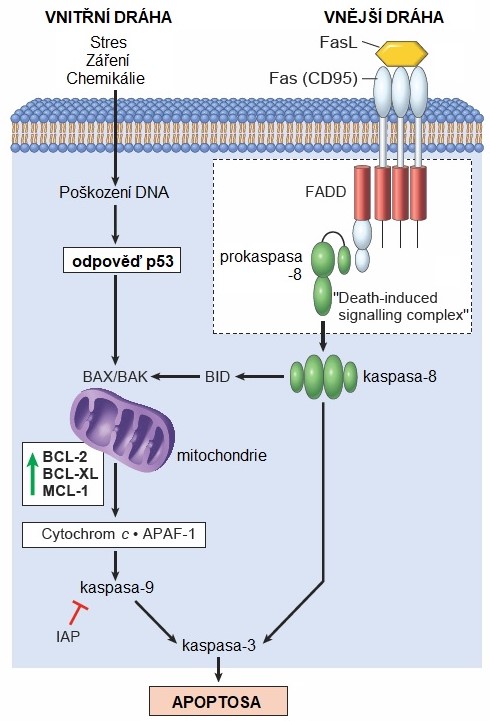
Obr. 6. Vnitřní a vnější cesta apoptosy (upraveno z Kumar a kol. 2014)
Angiogeneze
Angiogeneze (proces novotvorby krevních kapilár) je komplexní proces, který má zásadní význam pro růst nádoru, jeho invazivitu a metastázování. Angiogeneze je kontrolována rovnováhou mezi proangiogenními faktory (např. VEGF, bFGF, PDGF, TNFα, TGFβ, hormony – estrogeny, erythropoetin, NO, VCAM-1, E-selektin, cyklooxygenasa 2[6]) a angiogenními inhibitory (např. thrombospondin-1, interferon α/β, prolaktin, angiostatin, endostatin, vasostatin), u nádorů je tato rovnováha posunuta ve prospěch promotorů angiogeneze. Nádor (metastáza) potřebuje ke svému růstu přísun živin a kyslíku a odsun odpadních látek. Kromě toho jsou kapiláry v nádoru cestou, kterou se nádorové buňky dostávají do oběhu a mohou tak metastazovat. Pokud nádor nezíská schopnost novotvořit cévy, nemůže po dosažení velikosti zhruba 1-2 mm3 dále růst (cca 100-200 µm od cévy - překročení difuzního limitu pro kyslík). V počátku vývoje nemá většina lidských nádorů schopnost tvořit cévy a pravděpodobně po léta zůstávají malé nebo in situ, dokud není toto stadium ukončeno tzv. angiogenním přepnutím a nezačnou se tvořit nové cévy. Jeho příčinou je jednak zvýšená tvorba angiogenních faktorů nebo ztráta inhibitorů angiogeneze vlastními nádorovými buňkami, buňkami zánětu nebo dalšími buňkami nádorového stromatu, a tvorba proteas nádorovými buňkami nebo buňkami stromatu. Relativní nedostatek kyslíku díky hypoxii stabilizuje transkripční faktor HIF1α[7], který po translokaci do jádra aktivuje transkripci proangiogenních cytokinů VEGF a bFGF. Tyto faktory vytváření angiogenní gradient, který stimuluje proliferaci buněk endotelu a řídí růst nových cév k nádoru. Při novotvorbě cév nejprve endoteliální buňky stávajících cév proteolyticky rozloží bazální membránu pomocí plasminu a matrix metalloproteinas (MMP, některé typy proteolyticky aktivovány plasminem) a zahájí migraci do stromatu sousedních tkání, kde následně proliferují.
Mutace zasahující tumor-supresorové geny nebo onkogeny u nádorů může posunout rovnováhu ve prospěch angiogeneze. Ve zdravých buňkách p53 stimuluje expresi antiangiogenních faktorů (trombospondin-1) a brání expresi proangiogenních molekul (VEGF). V nádorových buňkách s mutovaným p53 tak dojde k posílení angiogeneze. Transkripce VEGF je ovlivněna též signály z RAS-MAP kinasové dráhy, takže v případě mutací genů ras a myc je exprese VEGF upregulována. U velkého množství pacientů s nádorovými onemocnění lze detekovat zvýšené hladiny VEGF a bFGF v séru a moči.
Invazivita a vznik metastáz
Invazivita a tvorba metastáz jsou výsledkem komplexních interakcí mezi nádorovými buňkami a normálním stromatem a jsou hlavní příčinou morbidity a mortality nádorových onemocnění. Metastázováním nádoru rozumíme jeho šíření do oblastí anatomicky vzdálených primárnímu ložisku. Tvorba metastáz velmi úzce souvisí se schopností tvořit nové cévy. Faktory, které se uplatňují v angiogenezi se více či méně podílejí i na tvorbě metastáz. Metastázování je složitý vícestupňový proces, který probíhá ve čtyřech etapách označovaných jako metastatická kaskáda: invaze – transport – nidace – růst metastázy.
První etapou metastatické kaskády je invaze nádoru do okolí, během níž dochází k průniku nádorových buněk extracelulární matrix (ECM) okolní tkáně a dále přes bazální membránu endotelu do lumina cévy. Invaze do okolí začíná uvolněním nádorových buněk díky výrazné/úplné ztrátě adheze vlivem inaktivující mutace nebo snížené exprese membránového glykoproteinu E-kadherinu, který se významně podílí na udržení pevných intercelulárních adhezí. Mutace E-kadherinu se velmi často vyskytuje u pacientů s různými typy epiteliálních nádorů (např. karcinom prsu, tlustého střeva, ovaria, žaludku). Ztráta adhezivity a zvýšení motility nádorových buněk je jedním z charakteristických rysů přeměny epiteliálních nádorových buněk na mezenchymovální fenotyp. Druhým krokem invaze je překonání bazální membrány ECM. Rozklad ECM je zprostředkován proteolytickými enzymy MMP, které působí na bazální membránu ECM i bazální membránu endotelií a usnadňují tak angiogenezi. MMP secernují buď samotné nádorové buňky, nebo buňky nádorového stromatu, které jsou indukovány růstovými faktory zachycenými na ECM. Rovněž rozkladné produkty kolagenu mají chemotaktické, angiogenní a prorůstové účinky.
Druhou etapou metastatické kaskády je transport nádorových buněk krevní nebo lymfatickou cestou. Jedná se o pasivní transport. Samotný průnik do cirkulace neznamená automaticky i tvorbu metastáz, protože nádorové buňky jsou náchylné k destrukci a transport z nich přežije jen asi 0,1 %. Nádorové buňky jsou eliminovány různými mechanismy, jedná se o mechanické faktory (tření, turbulence krevního proudu atd.), apoptosu indukovanou ztrátou adheze (tzv. anoikis) a imunitní mechanismy vrozené i adaptivní imunity a tzv. kyslíkový efekt. Nádorové buňky mají v cirkulaci tendenci se shlukovat mezi sebou nebo s krevními buňkami, zejm. trombocyty. Vznik shluků nádorových buněk s destičkami může zvýšit přežití nádorových buněk a jejich implantabilitu.
Nidace nádorových buněk a jejich průnik do tkáně, který probíhá především v kapilárách parenchymatózních orgánů, je třetí etapou metastatické kaskády. Adheze shluků tumor-destičky k endotelu i vzájemné interakce buněk jsou zprostředkovány pomocí cytoadhezivních molekul (CAMs, cell adhesion molecules), zejm. selektin E (interakce mezi buňkami ve shluku) a molekuly ze skupiny imunoglobulinů ICAM-1/2[8] a VCAM-1 (aktivace endotelu a adheze mikrotrombů k endotelu). Z těchto shluků se uvolňuje tromboxan A, který vyvolá ireverzibilní agregaci a tedy i fixaci mikrotrombu s nádorovými buňkami na stěnu cévy. Následuje průnik nádorových buněk bazální membránou zprostředkovaný MMP, angiogenními faktory a dalšími tkáňovými působky. Proliferace buněk mikrometastázy je vyvolána působením PDGF.
Metastatická kaskáda je završena růstem metastáz v novém prostředí. Ten ovšem neprobíhá uniformně. Buňky v metastáze zůstávají obvykle dlouhou dobu v klidovém stavu, aniž by však ztratily svůj proliferační potenciál, jen výjimečně diferencují. Proliferace buněk v metastáze závisí na přítomnosti růstových faktorů a na jejich poměru s faktory, které proliferaci brzdí. Na proliferaci metastáz se podílejí PDGF a autokrinní humorální faktory, dále produkty onkogenů (c-myc, c-erb, c-sic), které mají aktivitu růstových faktorů (FGF, TGF-α, EGF[9]). Proliferace je rovněž podpořena inhibicí apoptosy v nádorových buňkách. Základním předpokladem pro růst metastáz je plynulý přísun živin a kyslíku, proto je pro růst metastáz nezbytná schopnost angiogeneze.
[1] GADD45 = Growth Arrest and DNA Damage-inducible 45
[2] BAX = Bcl2-Associated Protein X (Bcl-2 = B-Cell Lymphoma-2)
[3] ATM = Ataxia Teleangiectasia Mutated; ATR = Ataxia Teleangiectasia and Rad3-Related
[4] TNF = Tumor Necrosis Factor; TRAIL = TNF-Related Apoptosis-Inducing Ligand; FasL = First Apoptosis Signal Ligand
[5] DR = Death Receptor; Fas = First Apoptosis Signal; TNFR = TNF Receptor
[6] VEGF = vaskulární endoteliální růstový faktor; bFGF = bazický fibroblastový růstový faktor; PDGF = růstový faktor destiček; TGFβ = transformující růstový faktor β; VCAM-1 = vascular cell adhesion molecule
[7] HIF1α = Hypoxia Inducible Factor 1α
[8] ICAM = Intercellular Adhesion Molecule
[9] EGF = epidermální růstový faktor