Poruchy hormonů kůry nadledvin
Kortisolismus
Jak je zřejmé z příkladu v Obr. 31, zobrazujícím regulaci kortisolu, porucha hormonální osy hypothalamus-hypofýza-nadledviny může mít příčinu v některé ze tří úrovní. Výsledné projevy mohou být podobné. Diagnostikují se biochemickým vyšetřením koncentrace hormonů v krvi. Léčba se pak podle lokalizace poruchy liší.
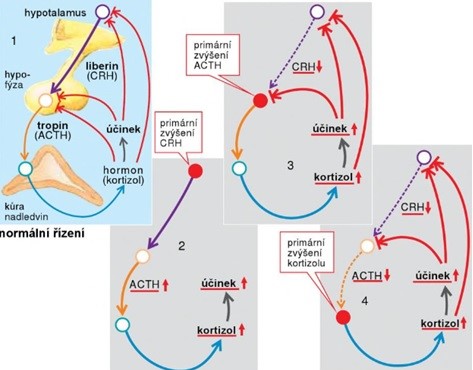
Obr. 31 Hormonální poruchy osy hypothalamus – hypofýza – nadledviny (podle Silbernagl a Lang 2012).
Poměrně časté jsou afunkční adenomy adenohypofyzární tkáně. Příznaky jsou v tomto případě způsobeny většinou anatomickou expanzí hypofyzární tkáně. Vznikají bolesti hlavy, poruchy zorného pole (útlak chiasma opticum) nebo parézy okohybných nervů. Teprve velké afunkční tumory způsobí snížení endokrinních funkcí adenohypofýzy. Hormonálně aktivní hyperplazie, adenomy nebo karcinomy hypofýzy se projeví v nadprodukci některého hypofyzárního hormonu s příslušnými endokrinními příznaky.
Hyperkortisolismus
Termíny Cushingův syndrom a Cushingova nemoc i podstata těchto nemocí jsou známy díky svému objeviteli americkému lékaři Harveyovi Cushingovi téměř sto let. Cushingův syndrom je název pro klinické příznaky a fyzické příznaky vyvolané nadbytkem glukokortikoidů.
- Periferní Cushingův syndrom (ACTH-independentní, primární)
Příčinou bývá endokrinně aktivní hyperplazie nebo nádor kůry nadledvin, který může být benigní (adenomy) nebo maligní (karcinomy).
- Centrální Cushingův syndrom (ACTH-dependentní, sekundární)
Nadbytek kortikoidů je v tomto případě způsoben endokrinně aktivním hypofyzárním nádorem se zvýšenou sekrecí adrenokortikotropního hormonu (kortikotropin, ACTH). Kortikotropin pak stimuluje kůru nadledvin, což má za následek oboustrannou adrenokortikální hyperplazii se zvýšenou produkcí kortikoidů. Tento typ se někdy označuje jako Cushingova nemoc.
Dříve se přísně rozlišovala Cushingova nemoc (z hypofyzární nadprodukce) a Cushingův syndrom (příčina je v nadledvinách). Dnes se již na tomto způsobu rozlišení netrvá.
- Ektopický Cushingův syndrom
Objevuje se při malobuněčném plicním karcinomu nebo při bronchiálním karcinoidu s tzv. paraneoplastickou tvorbou ACTH. Kortikotropin pak opět stimuluje produkci kortisteroidů v nadledvinách.
- Iatrogenní Cushingův syndrom
Vzhledem k tomu, že léčba chorob kortikoidy je velmi častá, syndrom může být navozen jejich nadměrnými dávkami.
Klinický obraz různých typů Cushingova syndromu je v podstatě stejný a je způsoben vysokou koncentrací kortikosteroidů. Typický je „centrální“ typ obezity s tukovými polštáři rozloženými na trupu a relativně tenkými končetinami. Obličej je zakulacený (MĚSÍČKOVINÝ OBLIČEJ), s tukovými polštáři kolem krku, typická je svalová atrofie, hypertenze, diabetes (z převahy kortisolu jako antagonisty insulinu), osteoporóza (Obr. 32).
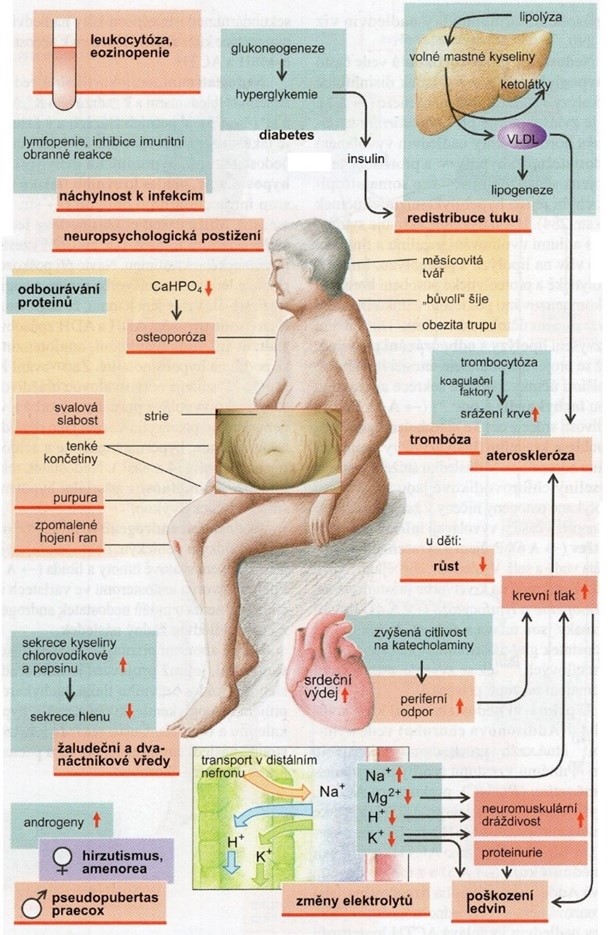
Obr. 31 Hyperkortisolismus – Cushingův syndrom (podle Silbernagl a Lang 2012)
Právě v diagnostice Cushingova syndromu je důležité biochemické laboratorní vyšetření. Vysoká koncentrace ACTH ukazuje na centrální nebo ektopickou příčinu, přičemž koncentrace kortisolu je vysoká vždy. Může se sledovat diurnální rytmus sekrece kortisolu. U zdravého člověka je typický pokles večer a v noci a zvýšený nástup koncentrace v časných ranních hodinách („ranní injekce kortisolu“ před očekávaným denním stresem), u nemocného s Cushingovým syndromem je koncentrace kortisolu vysoká i v noci). Provádí se také „dexamethasonový test“, který je ukázkou negativní zpětnovazebné regulace hormonů. U zdravého člověka vede podání syntetického glukokortikoidu dexamethasonu k potlačení sekrece kortikotropinu a tím následně i kortisolu. U pacientů s Cushingovým syndromem (bez ohledu na příčinu) se negativní zpětná vazba neprojeví. U periferního typu je sekrece kortisolu zcela autonomní a produkce kortisolu po podání dexamethasonu neklesá, v případě centrálního typu po vysokých dávkách dexamethasonu kortisol klesá (Tab. 5).
Tabulka 5. Základní laboratorní diferenciální diagnostika Cushingova syndromu

Hypokortisolismus
Addisonova choroba
Při hypokortisolismu bývá nedostatek jak glukokortikoidů, tak mineralokortikoidů, důvodem je většinou poškození kůry nadledvin, příčina však může spočívat i v poruše hypothalamu nebo adenohypofýzy. Proto se rozlišuje centrální a periferní forma Addisonovy choroby.
- Centrální (sekundární) Addisonova choroba
Je to méně častá choroba, příčinou bývá nádor hypothalamu nebo hypofýzy, případně poranění nebo infekce mozku. Kůra nadledvin je v tomto případě morfologicky i funkčně zcela v pořádku, je však nedostatečná signalizace řídícím hormonem. Vzhledem k tomu, že aldosteron je regulován jiným systémem (renin-angiotensin, viz výše), jeho produkce bývá zachována.
- Periferní (primární) Addisonova choroba
Příčina spočívá přímo v poškození kůry nadledvin. Tato forma je mnohem častější. V dnešní době je nejčastější příčinou autoimunní mechanismus. Tvoří se protilátky proti enzymům syntézy kortikoidů CYP21, CYP11A, CYP17 (viz Obr. 18). Zpravidla se zdůrazňuje postižení 21-hydroxylasy. Jinými příčinami může být relativně rychlá destrukce buněk kůry nadledvin např. meningokokem, kdy dojde ke krvácení do nadledvin s následným zničením buněk, dalšími příčinami mohou být metastázy nádorů, mykózy. Dříve bývala častější příčinou primární Addisonovy choroby tuberkulóza nadledvin.
U periferní formy bývá vzhledem k příčinám charakteristické postižení produkce jak glukokortikoidů, tak mineralokortikoidů. Kůra nadledvin má značné kapacitní rezervy v produkci kortikoidů a odhaduje se, že pro manifestaci choroby musí být zničeno 80‑90 % tkáně kůry nadledvin.
Vedle hypoglykemie a celkového neprospívání jsou u primárního aldosteronismu typické pigmentace např. v místech ohybu kůže, dásní, v místě útlaku prádlem (viz obr. 32). V důsledku nedostatku aldosteronu bývá hypotenze, hyponatrémie a hypokalémie.
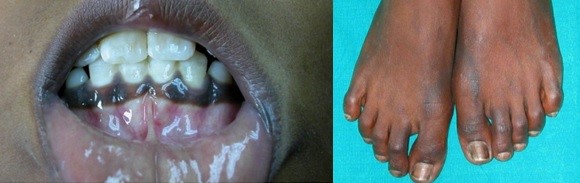
Obr. 33 Hyperpigmentace u Addisonovy choroby (Kumar et al. 2008)
Příčinou pigmentací je hypofyzární nadprodukce kortikotropinu (ACTH), vyvolaná nedostatkem negativní zpětné vazby při chybějícím kortisolu. Kortikotropin je syntetizován na základě exprese genu pro společný prohormon proopiomelanokortin. Vedle kortikotropinu jsou zároveň syntetizovány peptidy s melanotropním účinkem (viz obr. 34). Selektivní štěpení těchto peptidů je totiž až sekundární, po jejich syntéze. Celkový přehled změn u Addisonovy choroby ukazuje obr. 35.
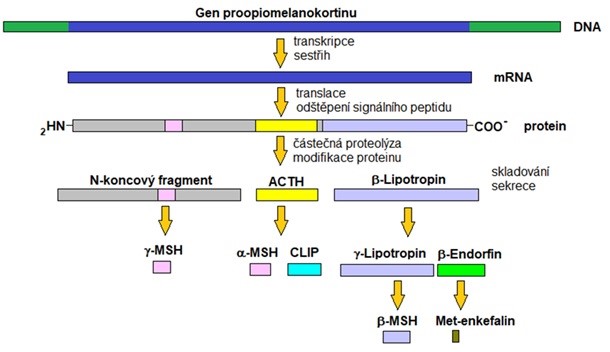
Obr. 34 Syntéza kortikotropinu (ACTH) ze společného prekurzoru proopiomelanokortinu (upraveno z Goodman 2009).
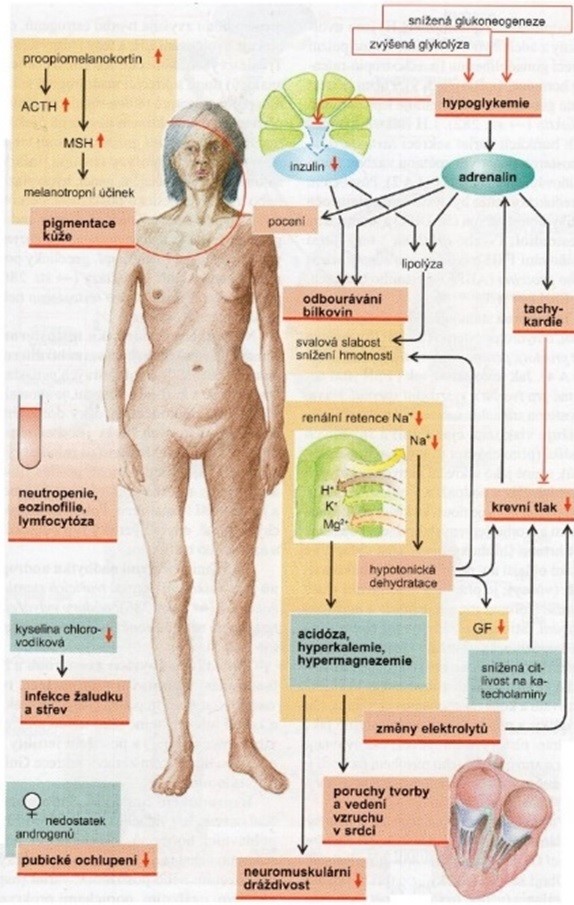
Obr. 35 Celkový přehled změn u Addisonovy choroby (podle Silbernagl a Lang 2012)
Aldosteronismus
Jako aldosteronismus se označují změny syntézy a působení mineralokortikoidu aldosteronu.¨
Hypoaldosteronismus
Snížení syntézy aldosteronu může být součástí výše popsaného celkového hypokortikalismu (Addisonova choroba), více či méně selektivního nedostatku aldosteronsynthasy (CYP11B) nebo mírné renální insuficience.
Hyperaldosteronismus
Zvýšená syntéza a sekrece aldosteronu může mít příčinu přímo v nadprodukci aldosteronu kůrou nadledvin (primární hyperaldosteronismus – Connův syndrom) nebo v aktivaci systému renin-angiotenzin-aldosteron (sekundární hyperaldosteronismus).
- Primární hyperaldosteronismus – Connův syndrom
Příčinou může být hyperplazie nebo adenom buněk zona glomerulosa kůry nadledvin.
- Sekundární hyperaldosteronismus
Může být způsoben snížení objemu cirkulující krve a hyperfunkcí buněk juxtaglomerulárního aparátu ledvin (zvýšení aktivity systému renin-angiotenzin-aldosteron), viz Obr. 35.
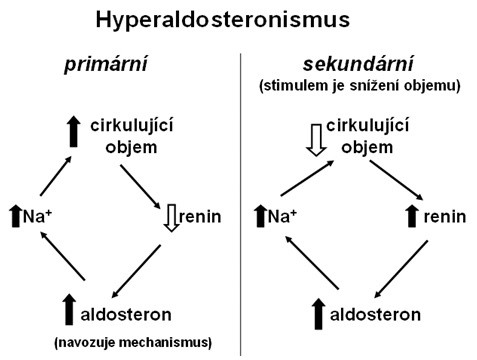
Obr. 36 Primární a sekundární hyperaldosteronismus
Příčiny změn hladin glukokortikoidů a mineralokortikoidů jsou shrnuty v Obr. 37.
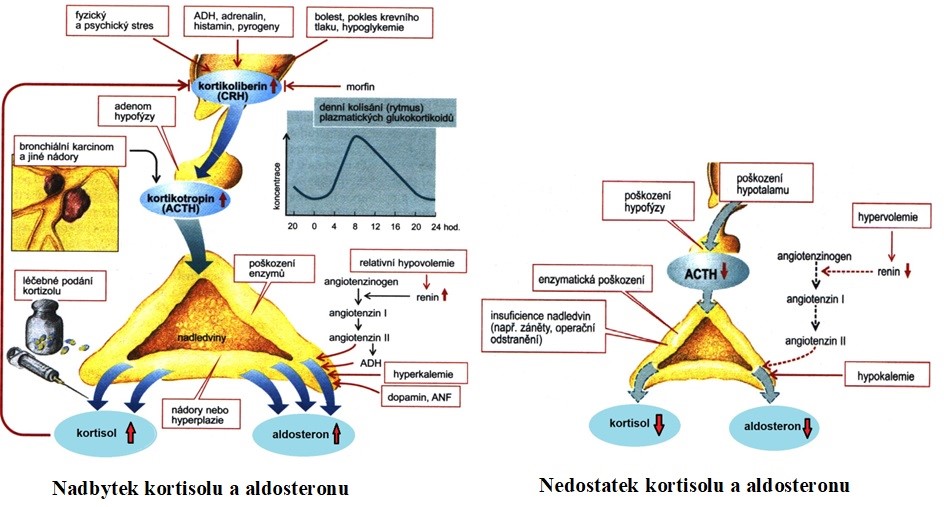
Obr. 37 Přehled příčin změn hladin glukokortikoidů a mineralokortikoidů (podle Silbernagl a Lang 2012)
Kongenitální adrenální hyperplazie
Pod název kongenitální adrenální hyperplazie (dříve adrenogenitální syndrom) patří skupina autosomálně recesivně dědičných poruch syntézy steroidních hormonů, jejichž příčinou je defekt některého z pěti nezbytných enzymů, katalyzujících jejich tvorbu. Enzymatický blok vede k deficitu příslušné části spektra steroidních hormonů za tímto defektem, zejména kortisolu. Nedostatkem kortisolu se odbrzdí negativní zpětná vazba, takže hypothalamus tvoří větší množství kortikoliberinu (CRH) a adenohypofýza secernuje větší množství kortikotropinu (ACTH). Ten vyvolává hyperplazii kůry nadledvin a stimuluje se tvorba těch steroidních hormonů, jejichž syntéza není blokována (Obr. 38).
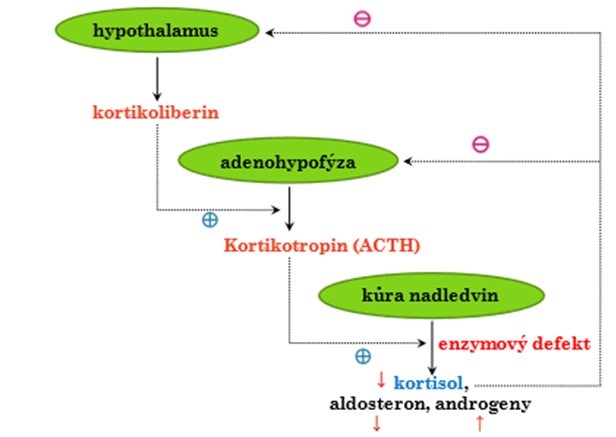
Obr. 38 Schema koncentračních změn kortikoidů při kongenitální adrenální hyperplazii.
Podle toho, jak je tímto způsobem nasměrována syntéza kortikoidů, objevuje se pro každý typ enzymového defektu specifický klinický obraz. Nejčastější typ kongenitální adrenákní hyperplazie je způsoben deficitem 21-hydroxylasy (CYP21, viz Obr. 39).
Klinický obraz kongenitální adrenální hyperplazie zahrnuje některé charakteristiky klinického obrazu výše popsaných hormonálních steroidních poruch, jako jsou hyponatremie, hyperkalemie, dehydratace, hypotenze, a zároveň obrazu z nadbytku nadledvinových androgenů (obr. 39 a tabulka 6; odtud starší název adrenogenitální syndrom).
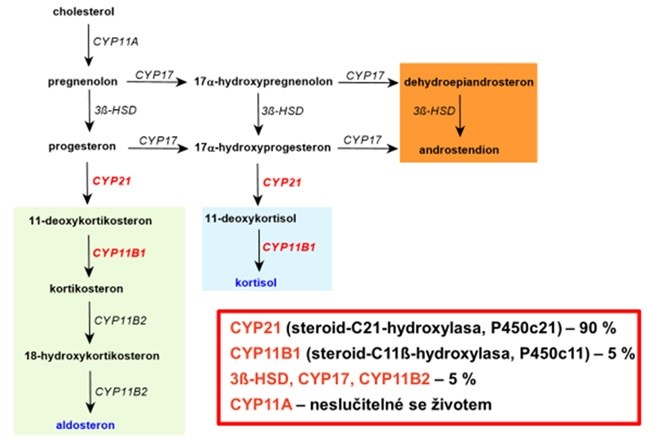
Obr. 39 Relativní poměr jednotlivých typů kongenitální adrenální hyperplazie
Tabulka 6 Charakteristika jednotlivých enzymových defektů syntézy kortikosteroidů
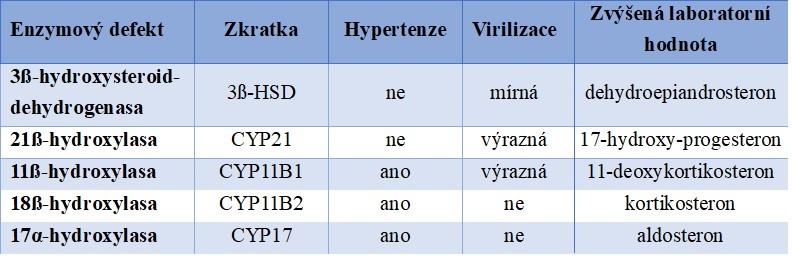
V případě deficitu 21-hydroxylasy se rozlišuje klasická (těžká) forma chroby a „neklasická“ lehčí forma (Tab. 7).
Tabulka 7 Deficit 21-hydroxylasy – dvě formy kongenitální adrenální hyperplazie

Syntéza estrogenů a její poruchy
Estrogeny jsou tvořeny z androgenů v jediném kroku za katalýzy enzymem aromatasou (viz Obr. 40).
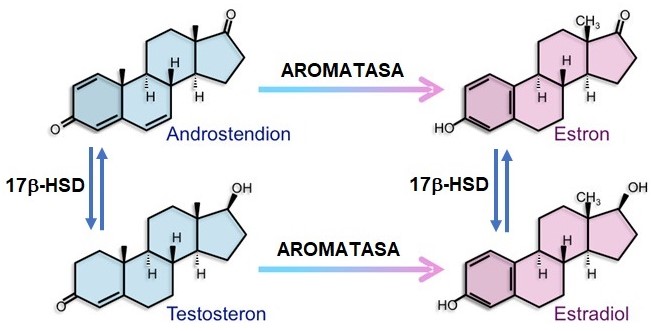
Obr. 40 Syntéza estrogenů z androgenů (upraveno z Rochira a Carani 2023)
Aromatasa (EC 1.14.14.14, CYP19A1) je enzym zodpovědný za klíčový krok v biosyntéze estrogenů, je proto důležitým faktorem v sexuálním vývoji. Katalyzuje poslední kroky biosyntézy estrogenů z androgenů (konkrétně přeměňuje androstendion na estron a testosteron na estradiol). Tyto kroky zahrnují tři postupné hydroxylace 19-methylové skupiny androgenů, následované současnou eliminací methylové skupiny jako formiátu a aromatizací A-kruhu. Aromatasa je monooxygenasa, patřící nadrodiny cytochromu P450 a v ní do skupiny enzymů, katalyzujících mnoho reakcí zapojených do steroidogeneze. Aromatasa je exprimována v gonádách, placentě, mozku, tukové tkáni, kostech a dalších tkáních. V játrech dospělého člověka je téměř nedetekovatelná. Aromatasa je však i ve tkáni karcinomu endometria, při endometrióze, v děložních myomech, při karcinomu prsu.
Aktivita aromatasy se zvyšuje s věkem, při obezitě a při konzumaci alkoholu. Její aktivita v tukové tkáni přispívá k obezitě u starších mužů (viz Obr. 41 a Obr. 42).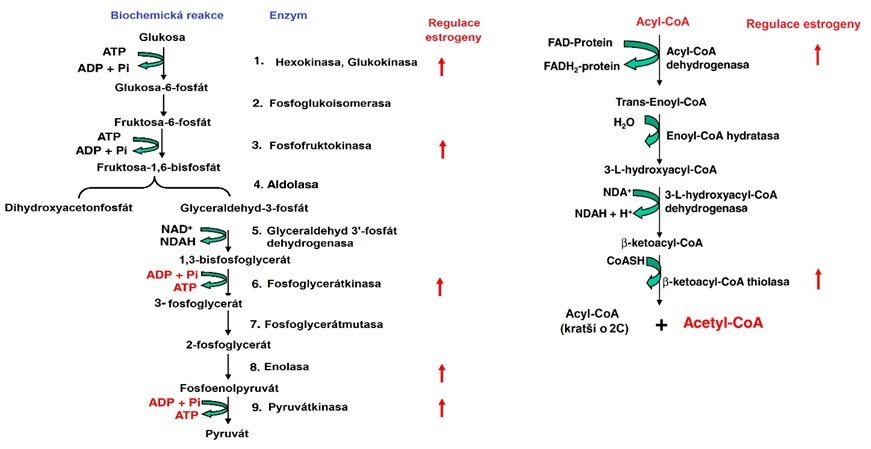
Obr. 41 Regulace glykolýzy (vlevo) a beta-oxidace (vpravo) estrogeny (upraveno z Chen et al. 2009).
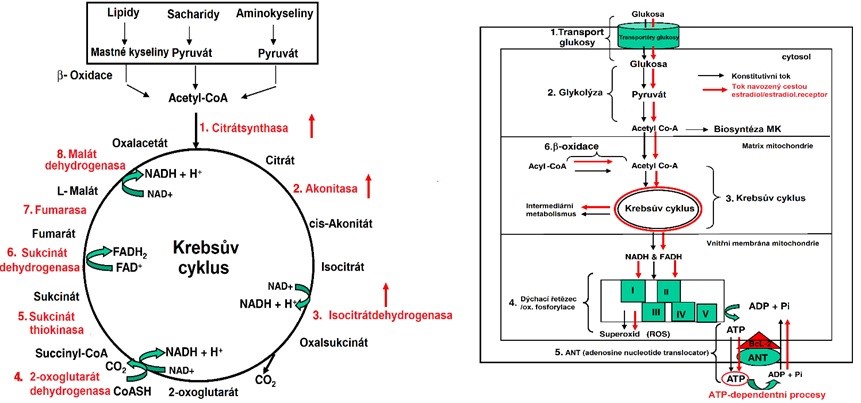
Obr. 42 Regulace citrátového cyklu (vlevo) a celkového toku energie estrogeny (upraveno z Chen et al. 2009). ANT (adenosine nucleotide translocator) zprostředkovává přenos ATP/ADP přes mitochondriální membránu, čímž transportuje většinu ATP generovaného MRC z mitochondrií výměnou za ADP a Pi. Rodina proteinů B-buněčného lymfomu 2 (Bcl-2) je považována za hlavní regulátor vnitřní dráhy apoptózy. Exprese Bcl-2 a některých izoforem ANT je stimulována estradiolem.
Vysoká exprese enzymu aromatasy v tukové tkáni zvyšuje přeměnu androgenů na estrogeny, což zase působí negativní zpětnou vazbu na hypotalamus a hypofýzu, inhibuje produkci gonadoliberinu (GnRH), luteinizačního hormonu (LH) a hormonu stimulujícího folikuly (FSH) a v důsledku toho klesá sekrece testosteronu ve varlatech, což vede k hypogonadotropnímu hypogonadismu.
Syndrom nedostatku aromatasy
Tento syndrom je způsoben mutací genu CYP19 a dědí se autosomálně recesivním způsobem. U ženy je příznakem primární amenorea. Jedinci obou pohlaví trpící tímto syndromem jsou vysocí, protože při nedostatku estrogenů se neuzavírají epifyzární štěrbiny. Nedostatečná aktivita aromatasy vede v těhotenství ke zvýšené koncentrace androgenů a virilizaci ženy.